Резюме
По данным Всемирной организации здравоохранения, в настоящее время около 422 млн людей во всем мире страдают сахарным диабетом. Стремительный рост заболеваемости сахарным диабетом привел к увеличению числа людей со сниженной трудоспособностью и повышению затрат на лечение. В 2016 г. 1,6 млн смертельных случаев произошло по причине сахарного диабета. Такой прирост инвалидности и смертности обусловлен осложнениями сахарного диабета: макро- и микроангиопатией, а также невропатией.
Одним из наиболее распространенных и тяжелых хронических осложнений диабета в настоящее время является диабетическая полиневропатия (ДПН). В 90% случаев ДПН осложняет течение сахарного диабета типа 1 и 2, становясь причиной снижения трудоспособности, инвалидизации и смертности пациентов.
Согласно современным представлениям, в патогенезе ДПН основную роль играют гипергликемия и высокий уровень гликированного гемоглобина. Однако механизмы развития ДПН полностью не раскрыты. Известно, что далеко не во всех случаях возникновение и прогрессирование ДПН можно объяснить традиционными факторами риска. Следовательно, поиск недостающих звеньев патогенеза, которые могут стать причиной возникновения и прогрессирования ДПН, остается чрезвычайно актуальной задачей.
В последние годы накопленные данные свидетельствуют о повышенном уровне гомоцистеина (гипер-гомоцистеинемия) как о новом факторе риска, усугубляющем протекание диабетических микро- и макрососудистых осложнений. Многие исследователи в своих наблюдениях показали корреляцию между гипер-гомоцистеинемией и степенью выраженности ДПН. Согласно данным отечественных и зарубежных авторов, причиной повышенного уровня гомоцистеина при сахарном диабете типа 2 может стать нарушение кобаламинового обмена, чему способствует прием такого сахароснижающего препарата, как метформин. Роль гипергомоцистеинемии в развитии диабетических осложнений, в том числе ДПН, заключается в активации оксидативного стресса. Кроме этого, под влиянием повышенного уровня гомоцистеина нарушается функция эндотелия. Оксидативный стресс и эндотелиальная дисфункция являются основными взаимосвязанными механизмами в развитии ДПН. В обзоре описаны патогенетические процессы, в ходе которых гипергомоцистеинемия нарушает окислительно-восстановительные процессы организма и функцию эндотелия, ухудшая течение ДПН.
С каждым годом число людей, страдающих сахарным диабетом (СД), стремительно увеличивается. По данным Всемирной организации здравоохранения, с 1980 по 2014 г. число заболевших возросло в 4 раза: со 108 до 422 млн человек. Стремительный рост заболевания стал причиной более 1,6 млн смертей в 2012 г., что связано с осложнениями СД: макроангиопатией, микроангиопатией и невропатией [1].
Одним из наиболее распространенных и тяжелых хронических осложнений СД, затрагивающих до 90% пациентов с СД типа 1 и 2, в настоящее время является диабетическая полиневропатия (ДПН) [2]. Согласно современным представлениям в патогенезе ДПН основную роль играют гипергликемия и высокий уровень гликированного гемоглобина [3]. Неблагоприятными прогностическими факторами для развития ДПН являются повышение уровней триглицеридов, липопротеинов низкой плотности и фибриногена в крови, а также возраст пациента, длительность СД, употребление алкоголя и курение [4]. Однако механизмы развития ДПН полностью не раскрыты. Известно, что далеко не во всех случаях возникновение и прогрессирование ДПН можно объяснить традиционными факторами риска. Следовательно, поиск недостающих звеньев патогенеза остается чрезвычайно актуальной задачей. В последние годы накопленные данные указывают на новый фактор риска, поражающий нервную систему, – повышенный уровень гомоцистеина (гипергомо-цистеинемия).
Многие авторы принимают гипергомоцистеинемию как независимый фактор риска, осложняющий течение целого ряда сердечно-сосудистых и нейродегенеративных процессов [5]. Не вызывает сомнение, что гипергомоцистеинемия вносит большой вклад в сопровождение и усугубление протекания диабетической нефропатии [6], ретинопатии [7] и макрососудистых осложнений [8]. Механизмы проявления таких патологических эффектов гомоцистеина и продукта его аутоокисления – гомоцистеиновой кислоты многообразны. Хотя нет крупномасштабных исследований, многие зарубежные и отечественные авторы в своих наблюдениях выявили, что гомоцистеин обладает прямым и опосредованным, за счет активации оксидативного стресса и снижения уровня монооксида азота, поражающим действием на эндотелий сосудов и нервную ткань [9-12]. При этом даже кратковременная инкубация клеток с гомоцистеиновой кислотой, в концентрациях сравнимых с наблюдаемыми при гипергомоцистеинемии, индуцирует развитие апоптоза. Следовательно, гипергомоцистеинемия у пациентов с СД типа 2 за счет активации взаимосвязанных патогенетических механизмов – оксидативного стресса и эндотелиальной дисфункции, которые лежат в основе развитии диабетических осложнений, может стать причиной прогрессирования ДПН.
Причина гипергомоцистеинемии при сахарном диабете типа 2
Гомоцистеин – серосодержащая аминокислота, возникающая в ходе метаболизма незаменимой аминокислоты метионина. После образования из метионина гомоцистеин реме-тилируется за счет извлечения одноуглеродного фрагмента из аминокислоты серина с образованием метионина или вступает в реакцию транссульфирования с превращением в нетоксичную аминокислоту цистеин. Реметилирование и транссульфирование, являясь основными путями утилизации гомоцистеина, осуществляются ферментами, активность которых зависит от наличия витаминов В6, В12 и активной формы фолиевой кислоты. В процессе транссульфирования из гомоцистеина образуется цистеин – предшественник антиоксидантного пептида глутатиона (GSH) [13]. Реметилирование гомоцистеина осуществляется под действием метионинсинтетазы, коферментом которого является циан-кобаламин (витамин В12). В результате образуется метионин, который затем превращается в S-аденозилметионин (SAM). SAM служит в качестве донора для нескольких реакций с участием ДНК, РНК и белков [14] (рис. 1).
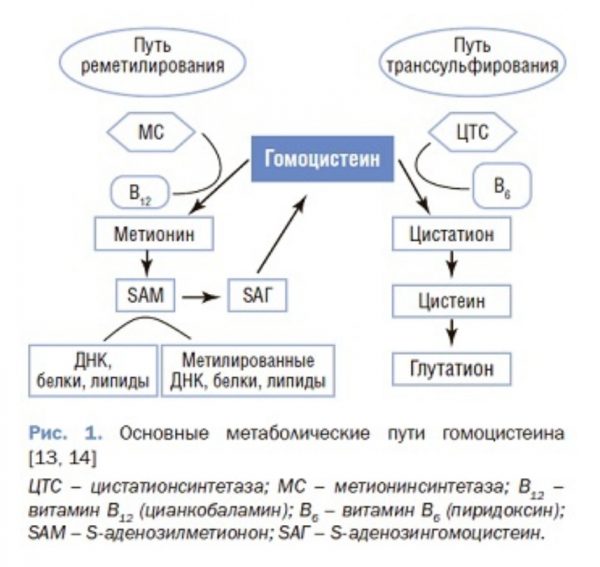
Совокупность этих процессов обеспечивает низкий уровень гомоцистеина в крови. В физиологических условиях основная масса гомоцистеина в крови связана с белками или цистеином, только его незначительная часть находится в свободном виде. Свободный гомоцистеин очень быстро окисляется в гомоцистеиновую кислоту. Все эти формы определяются при клинических анализах крови в виде “общего гомоцистеина”. У здоровых людей концентрация гомоцистеина в плазме крови составляет 5-15 мкмоль/л. Термин “гипергомоцистеинемия” используют тогда, когда уровень гомоцистеина выше 15 мкмоль/л [15]. Из многочисленных причин нарушения утилизации гомоцистеина при СД типа 2 наиболее важно нарушение кобаламинового обмена, которое приводит к дефициту витамина В12. При этом блокируется один из основных путей метаболизма гомоцистеина – реметилирование, что приводит к гипергомоцистеинемии [9, 12, 16]. Еще важно отметить, что большое клинико-диагностическое значение для кобаламинового обмена имеют показатели гомоцистеина в плазме крови и/или моче, так как сывороточный уровень гомоцистеина чувствителен к раннему клеточному (метаболическому) дефициту витамина В12, и уровень гомоцистеина повышается раньше, чем снижается концентрация витамина В12 в сыворотке крови [17]. Это значит, что незначительный дефицит витамина В12, который протекает бессимптомно, может стать причиной клинически значимой гипергомоцистеинемии.
Многие авторы в разное время в своих исследованиях выявили нарушение кобаламинового обмена при СД типа 2 [18, 19], объясняя это явление приемом такого сахароснижающего препарата, как метформин. Это связано с действием метформина на моторику желудочно-кишечного тракта и на скорость всасывания глюкозы через стенку кишечника. Метформин, замедляя всасывание глюкозы из стенки кишечника, приводит к нарушению микрофлоры кишечника и мальабсорбции витамина В12[20]. Сывороточный уровень витамина В12 начинает снижаться через 3 мес от начала лечения метформином [21]. В настоящее время, согласно рекомендациям ADA, метформин является препаратом первой линии терапии СД типа 2 [22]. Его врачи рекомендуют более 50% пациентов [23]. Кроме этого, метформин предпочтительно комбинируют с другими сахароснижающими препаратами и с инсулином [24]. Учитывая распространенность СД типа 2 и широкое использование метформина, становится понятна степень выраженности дефицита витамина В12 у пациентов, получающих метформин [25].
Сам дефицит витамина В12 становится причиной прогрессирования ДПН, вызывая гипергомоцистеинемию по описанному выше механизму. И в зарубежной, и в отечественной литературе данные исследователей последних нескольких лет демонстрируют корреляцию между гипергомоцистеинемией и дефицитом витамина В12 с выраженностью ДПН.
В числе таких исследований работа D. Wile и соавт., где была показана статистическая значимая связь между уровнями гомоцистеина и витамина В12 с выраженностью ДПН у пациентов с СД типа 2, которые получали метформин. Исследование показало, что в группе пациентов с дефицитом витамина В12 и гипергомоцистеинемией клинические и электрофизиологические признаки ДПН более выражены и сильно коррелируют с дозой и продолжительностью терапии метформином [26]. Аналогичные результаты получили в своем исследовании R. Roy и соавт., показав корреляцию между дефицитом витамина В12 и гипергомоцистеинемией с нарушением нервной проводимости у пациентов с СД типа 2, принимающих метформин от 2 до 3 лет [27].
В своем исследовании M. Sahin и соавт. сравнили влияние метформина и росиглитазона на уровень витамина В12, фолатов и гомоцистеина у пациентов с СД типа 2. Снижение уровня фолатов и витамина В12 и повышение уровня гомоцистеина было более значимым в группе пациентов, принимающих метформин, по сравнению с группой пациентов, принимающих росиглитазон [28].
Таким образом, исходя из вышесказанного не вызывает сомнения, что гипергомоцистеинемия при СД может быть результатом нарушенного кобаламинового обмена, а причиной активации оксидативного стресса и нарушения функции эндотелия, кроме традиционных факторов риска СД, может стать повышенный уровень свободного гомоцистеина.
Влияние уровня гомоцистеина на активацию оксидативного стресса
Как считают многие авторы, активация оксидативного стресса в условиях гипергомоцистеинемии связана с усиленным образованием гомоцистеиновой кислоты. С одной стороны, гомоцистеин и гомоцистеиновая кислота непосредственно стимулируют рост активных форм кислорода (АФК) в цитоплазме нейронов, становясь причиной апоптоза нейронов [15, 29], а с другой – гомоцистеиновая кислота, вступая во взаимодействие с эндогенными тиолами, к которым относятся метионин, цистеин, таурин, глутатион, сероводород, липоевая кислота, полисульфиды и др., обеспечивающие окислительно-восстановительный баланс клеток, генерирует образование супероксид-анион-радикала (O.2),восстанавливаясь в гомоцистеин. Чрезмерное образование O.2стимулирует производство других АФК, в том числе перекиси водорода (H2O2) и гидроксильного радикала (ОН.), более токсичных, чем O.2. Необходимо отметить, что количество АФК как в норме, так и в условиях окислительного стресса регулируется экзогенно вводимыми веществами с антиоксидантной активностью (витамины А, С, Е, фолиеваякислота, глутатион, коэнзим Q10 и др.) и ферментами анти-оксидантной защиты: супероксиддисмутазы, глутатионпероксидазы, каталазы. Под влиянием супероксиддисмутазы из O.2 в реакции дисмутации образуется менее реакционноспособная Н2О2. В дальнейшем Н2О2 в реакции, катализируемой каталазой, распадается на инертные молекулыкислорода (О2) и воды (Н2О). Другой путь распада Н2О2 до воды происходит под действием системы ГПО при участии глутатиона (GSH) (рис. 2).
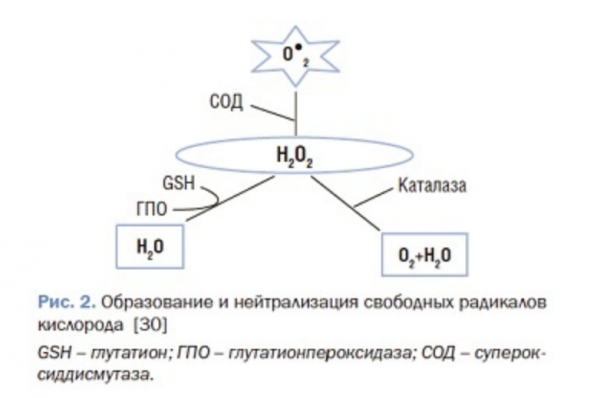
В норме антиоксидантная система работает как буфер, нейтрализуя свободные радикалы. А при патологических состояниях, когда усиливается образование АФК, запасы антиоксидантной системы истощаются, что становится причиной активации оксидативного стресса [30]. Одной из причин усиленного образования АФК является гипергомоцисте-инемия. В экспериментальных условиях было доказано, что субфизиологическая концентрация гомоцистеина (от 1 до 10 ммоль/л) также может привести к образованию активных форм кислорода [31]. Еще важно отметить, что гипергомоцистеинемия приводит к истощению внутриклеточных запасов антиоксидантной системы, таких как глутатионпероксидаза и глутатион, что становится причиной ослабления антиоки-дантной защиты организма [32].
Следовательно, накопление АФК в большом количестве и истощение запасов системы антиоксидантной защиты в условиях гипергомоцистеинемии необратимо повреждают клетки, сосуды микроциркуляторной системы [33] и нервные волокна [34]. Под влиянием АФК происходят удвоение базальной мембраны, пролиферация эндотелиальных клеток, пристеночное отложение фибрина, утолщение стенок и сужение просвета эндоневральных капилляров (vasa nevrorum). Эти изменения приводят к эндоневральной микроангиопатии, нарушению кровоснабжения и ишемии нерва [35-39]. Нарушение кровоснабжения в тканях приводит к гипоксии и еще большей генерации свободных радикалов. Таким образом, замыкается патологический круг образования свободных радикалов, что усугубляет морфофункциональное нарушение клеток и приводит к их гибели [40]. Наиболее уязвимой частью нервной клетки к действию патологических факторов является аксон. На начальных стадиях его поражения появляются признаки дегенерации тонких безмиелиновых волокон. В дальнейшем к этому процессу присоединяются истончение миелинового слоя, расширение эндоневрального пространства, повреждение шванновских клеток (леммоцитов), формирование участков демиелинизации и ремиелинизации [41, 42].
Влияние уровня гомоцистеина на функцию эндотелия сосудов
Как активация оксидативного стресса, так и нарушения функции эндотелия при СД происходят параллельно. Дополняя друг друга, оба процесса составляют основу патогенеза диабетических осложнений, в том числе ДПН.
Достаточно большой объем научной работы посвящен роли эндотелиальной дисфункции в развитии многих патологических процессов (нарушение регуляции сосудистого тонуса, функции свертываемой системы, кровоснабжения тканей организма и др.). Одна из главных функций эндотелия сосудов состоит в том, что он является важным регулятором гомеостаза организма. Клетки эндотелия, выстилая изнутри стенки всех сосудов, способны реагировать на механические и химические воздействия, протекающие в просвете сосудов крови. Эндотелий регулирует тонус сосудов и кровоток в тканях, активно вырабатывая вазоактивные вещества. Из них монооксид азота (N0) и простациклины обладают сосудорасширяющим эффектом, а сосудосуживающим – эндотелия-1 (ET-1), ангиотензин II и тромбоксан A2. Клетки эндотелия также вырабаты-вают анти- и прокоагулянты, обеспечивая текучесть крови [43, 44]. В физиологических условиях изнутри сосудистая стенка преобладает антикоагулянтным, сосудорасширяющим, антиоксидантным, противовоспалительным и антитромботическим эффектом [45]. Этому способствуют гладкая поверхность и положительный заряд эндотелия, а также синтез им соответствующих биологически активных веществ. Под влиянием повреждающих агентов (механических, инфекционных, обменных, иммунокомплексных и т.п.) резко меняется направление эндокринной активности эндотелия в противоположную сторону: усиливается образование коагулянтов и вазоконстрикторов, что препятствует кровопотере. При продолжительном воздействии патогенных факторов повреждаются клетки эндотелия, обнажается субэндотелиальный слой, что приводит к дисфункции эндотелия. В таких случаях эндотелий начинает играть ключевую роль в патогенезе ряда системных патологий (атеросклероз, гипертония, инсульты, инфаркты и др.), так как неоднократно увеличивается синтез оксидантов, вазоконстрикторов, агрегантов и тромбогенных факторов (рис. 3) [46].
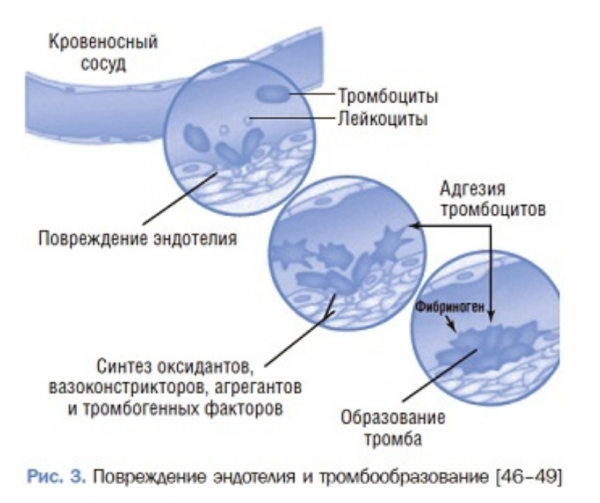
Несомненно, патогенез эндотелиальной дисфункции многофакторен, но многие авторы считают, что основным и общим механизмами, за которыми следует нарушение вазоактивного, воспалительного, гемостатического и окислительно-восстановительного гомеостаза в сосудистой системе организма, являются активация оксидативного стресса и истощение запасов системы антиоксидантной защиты. Нарушение окислительно-восстановительного баланса организма характеризуется повышением образования АФК. В последующем АФК, с одной стороны, блокируют активность NO-синтазы, а с другой – конъюгируясь с монооксидом азота (NO), непосредственно участвуют в образовании пероксинитрита, приводя к снижению биодоступности NO. В свою очередь NO является важным эндокринным вазорегулятором, модулирующим кровоток в микроциркуляции. Дисфункция эндотелия наблюдается, когда есть дисбаланс в производстве и потреблении NO. Снижение биодоступности NO создает благоприятные условия для активации тромбообразования и медиаторов воспаления, что в конечном итоге приводит к структурному повреждению эндотелиальных клеток. Еще одной причиной повреждения эндотелиальных клеток является высокий уровень пероксинитрита, который образуется в ходе воздействие АФК с NO. Пероксинитрит приводит к гибели клеток организма, так как обладает цитотоксическим эффектом и повреждает ДНК, липиды и белки.
Таким образом, дисфункция эндотелия, нарушая кровоснабжение в органах за счет усиления тромбообразования, вазоконстрикции в тканях всего организма, усиливает оксидативный стресс [9, 47-49]. Замыкается патологический круг, что еще больше усугубляет нарушение гомеостаза организма.
Одним из таких повреждающих эндотелий факторов, который нарушает его функцию, является гомоцистеин. Исследователи в своих наблюдениях выявили, что высокий уровень гомоцистеина коррелирует с повышенным риском развития сосудистых заболеваний [50]. Дальнейшие эпидемиологические исследования доказали, что гипергомо-цистеинемия ассоциируется с повреждением сердечнососудистой системы, независимо от других факторов риска. Многие авторы считают, что возможными механизмами, с помощью которых гомоцистеин вызывает поражение сосудов, являются повреждение эндотелия за счет снижения биодоступности монооксида азота, подавление активности антиоксидантных ферментов и стимуляция воспаления из-за активации оксидативного стресса [29, 51, 52].
Связь гипергомоцистеинемии и эндотелиальной дисфункции была доказана как in vitro, так и in vivo. Исследования показали, что гипергомоцистеинемия вызывает непосредственное патологическое влияние на эндотелий сосудов. В исследовании K. Woo и соавт. приняли участие здоровые добровольцы в возрасте 53±9 лет. Их разделили на 2 группы: с высоким (34,8±8,5 мкмоль/л) и нормальным (9,9±3,2 мкмоль/л) уровнем гомоцистеина. Эндотелий-зависимая дилатация сосудов в группе с гипергомоцистеинемией была ниже (6,5±1,7%) по сравнению с контрольной группой (10,8±1,7%) (p<0,001) [53]. A. Tawakol и соавт. показали, что эндотелий-зависимая вазодилатация была значительно ухудшена у пациентов с гипергомоцистеинемией по сравнению с контрольной группой (3,7±0,6 против 8,1±1,2%, p=0,004), тогда как эндотелий-независимая вазодилатация значимо не отличалась (10,1±1,6 против 9,3±1,5%, p=NS) [54]. По мнению Bellamy, гипергомоцистеинемия, вызванная увеличенным употреблением метионина и белка в пище, приводит к нарушению эндотелий-зависимой вазодилатации и эндотелиальной дисфункции [55, 56].
Заключение
Исходя из вышеизложенного гипергомоцистеинемия непосредственно и опосредованно, за счет активации оксидативного стресса, приводит к поражению нейронов, нервных волокон и эндотелия мелких сосудов (таких как vasa nevrorum), осложняя течение ДПН. Управление уровнем гомоцистеина в крови у пациентов с СД типа 2, принимающим метформин, имеет большое клинико-диагностическое значение в профилактике и лечении ДПН.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Литература
1. Global report on diabetes of WHO. 2016.
2. Tesfaye S., Boulton A.J., Dickenson A.H. Mechanisms and management of diabetic painful distal symmetrical polyneuropathy // Diabetes Care. 2013. Vol. 36. P. 2456-2465.
3. The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of Diabetes on the development and progression of long-term complication in insulin-dependent diabetes mellitus // N. Engl. J. Med. 1993. Vol. 329. P. 977-986.
4. Maser R.E., Steenkiste A.R., Dorman J.S. et al. Epidemiological correlates of diabetic neuropathy: report from Pittsburgh Epidemiology of Diabetes Complications Study // Diabetes. 1989. Vol. 38, N 11. P. 1456.
5. Wang H., Cui K., Xu K., Xu S. Association between plasma homocysteine and progression of early nephropathy in type 2 diabetic patients // Int. J. Clin. Exp. Med. 2015. Vol. 8, N 7. P. 11 174-11 7 80.
6. Li J., Shi M., Zhang H., Yan L. et al. Relation of homocysteine to early nephropathy in patients with type 2 diabetes // Clin. Nephrol. 2012. Vol. 77, N 4. P 305-310.
7. Brazionis L., Rowley K., Itsiopoulos C., Harper C.A. et al. Homocysteine and Diabetic Retinopathy // Diabetes Care. 2008. Vol. 31, N 1. P. 50-56.
8. Rudy A., Kowalska I., Strqczkowski M., Kinalska I. Homocysteine concentrations and vascular complications in patients with type 2 diabetes // Diabetes Metab. 2005. Vol. 31, N 2. P 112-117.
9. Кичигина О.Н., Голубева Т.И., Трошина И.А., Романова Н.В. Патогенетическое значение гипергомоцистеинемии при неалкогольной жировой болезни печени // Мед. наука и образование Урала. 2015. № 3. С. 179.
10. Obeid R., Herrmann W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia // FEBS Lett. 2006. Vol. 580, N 13. P 2994-3005.
11. Poddar R., Paul S. Novel crosstalk between ERK MAPK and p38 MAPK leads to homocysteine-NMDA receptor mediated neuronal cell death // J. Neurochem. 2013. Vol. 124, N 4. P 558-570.
12. Skovierova H., Vidomanova E., Mahmood S., Sopkova J. et al. The molecular and cellular effect of homocysteine metabolism imbalance on human health // Int. J. Mol. Sci. 2016. Vol. 17, N 10. Article ID e1733.
13. Cao Y., Chai J.G., Chen Y.C., Zhao J. et al. Beneficial effects of danshensu, an active component of salvia miltiorrhiza, on homocysteine metabolism via the trans-sulphuration pathway in rats // Br. J. Pharmacol. 2009. Vol. 157. P. 482-490.
14. Selhub J. Homocysteine metabolism // Ann. Rev. Nutr. 1999. Vol. 19. P. 217-246.
15. Болдырев А.А. Почему токсичен гомоцистеин? // Природа. 2009. № 10 (1130). С. 18-23.
16. Perta-Kajan J., Jakubowski H. Paraoxonase 1 and homocysteine metabolism // Amino Acids. 2012. Vol. 43. P 1405-1417.
17. England J.D., Gronseth G.S., Franklin G., Carter G.T. et al.; American Academy of Neurology. Practice parameter: evaluation of distal symmetric polyneuropathy: role of laboratory and genetic testing (an evidence-based review). Report of the American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, and American Academy of Physical Medicine and Rehabilitation // Neurology. 2009. Vol. 72. P 177-184.
18. Ting R.Z., Szeto C.C., Chan M.H., Ma K.K. et al. Risk factors of vitamin B12 deficiency in patients receiving metformin // Arch. Intern. Med. 2006. Vol. 166. P. 1975-1979.
19. Aroda V.R., Edelstein S.L., Goldberg R.B. et al.; Diabetes Prevention Program Research Group. Long-term metformin use and vitamin B12 deficiency in the Diabetes Prevention Program Outcomes Study // J. Clin. Endocrinol. Metab. 2016. Vol. 101. P 1754-1761.
20. Berchtold P., Bolli P., Arbenz U., Keiser G. Disturbance of intestinal absorption following metformin therapy (observations on the mode of action of biguanides) // Diabetologia. 1969. Vol. 5. P 405-412.
21. Buvat D.R. Use of metformin is a cause of vitamin B12 deficiency // Am. Fam. Physician. 2004. Vol. 69. P 264.
22. American Diabetes Association. 8. Pharmacologic approaches to glycemic treatment // Diabetes Care. 2017. Vol. 40, N 1. P 64-74.
23. Turner L.W., Nartey D., Stafford R.S., Singh S. et al. Ambulatory treatment of type 2 diabetes in the U.S., 1997-2012 // Diabetes Care. 2014. Vol. 37, N 4. P 985-992.
24. Nathan D.M., Buse J.B., Davidson M.B., Heine R.J. et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes // Diabetes Care. 2006. Vol. 29. P 1963-1972.
25. Zalaket J., Wehbe T., Jaoude E. Vitamin B12 deficiency in diabetic subjects taking metformin: a cross sectional study in a Lebanese cohort // J. Nutr. Intermediary Metab. 2018. Vol. 11. P 9-13.
26. Wile D.J., Toth C. Association of metformin, elevated homocysteine, and methylmalonic acid levels and clinically worsened diabetic peripheral neuropathy // Diabetes Care. 2010. Vol. 33, N 1. P 156-161.
27. Roy R.P., Ghosh K., Ghosh M., Acharyya A. et al. Study of vitamin B 12 deficiency and peripheral neuropathy in metformin-treated early type 2 diabetes mellitus // Indian J. Endocr. Metab. 2016. Vol. 20. P 631637.
28. Sahin M., Tutuncu N.B., Ertugrul D., Tanaci N. et al. Effects of metformin or rosiglitazone on serum concentrations of homocysteine, folate, and vitamin B12 in patients with type 2 diabetes mellitus // J. Diabetes Complications. 2007. Vol. 21. P 118-123.
29. Pop-Busui R., Sima A., Stevens M. Diabetic neuropathy and oxidative stress // Diabetes Metab. Res. Rev. 2006. Vol. 22, N 4. P 257-273.
30. Аметов А.С., Камынина Л.Л., Рождественская О.А., Пашкова Е.Ю. Положительные метаболические и антиоксидантные свойства тестостерон-заместительной терапии при сочетании сахарного диабета типа 2 и гипогонадизма // Эндокринология: новости, мнения, обучение. 2016. № 3. С. 83-93.
31. Jacobsen D.W. Hyperhomocysteinemia and oxidative stress // Ar-terioscler. Thromb. Vasc. Biol. 2000. Vol. 20. P 1182-1184.
32. Weiss N., Zhang Y.Y., Heydrick S., Bierl C. et al. Overexpression of cellular glutathione peroxidase rescues homocyst(e)ine-induced endothelial dysfunction // Proc. Natl Acad. Sci. USA. 2001. Vol. 98, N 22. P 12 503-12 508.
33. Creager M.A., Luscher T.F., Cosentino F., Beckman J.A. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: part I // Circulation. 2003. Vol. 108. P 1527-1532.
34. Brownlee M. Biochemistry and molecular cell biology of diabetic complications // Nature. 2001. Vol. 414, N 6865. P 813-820.
35. Malik R.A., Tesfaye S., Thompson S.D., Veves A. et al. Endoneurial localisation of microvascular damage in human diabetic neuropathy // Diabetologia. 1993. Vol. 36. P 454-459.
36. Malik R.A., Newrick P.G., Sharma A.K., Jennings A. et al. Microangiopathy in human diabetic neuropathy: relationship between capillary abnormalities and the severity of neuropathy // Diabetologia. 1989. Vol. 32, N 2. P. 92-102.
37. Cameron N.E., Eaton S.E., Cotter M.A., Tesfaye S. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy // Diabetologia. 2001. Vol. 44. P. 1973-1988.
38. Tuck R.R., Schmelzer J.D., Low P.A. Endoneurial blood flow and oxygen tension in the sciatic nerves of rats with experimental diabetic neuropathy // Brain. 1984. Vol. 107, N 3. P. 935-950.
39. Yagihashi S., Mizukami H., Sugimoto K. Mechanism of diabetic neuropathy: where are we now and where to go? // J. Diabetes Investig. 2011. Vol. 2, N 1. P. 18-32.
40. Недосугова Л.В. Диабетическая полинейропатия и окислительный стресс. Патогенез, диагностика, лечение : учебное пособие. М., 2015. С. 54-55.
41. Schreiber A.K., Nones C., Reis R.C., Chichorro J.G. et al. Diabetic neuropathic pain: physiopathology and treatment // World J. Diabetes. 2015. Vol. 6, N 3. P 432-444.
42. Jelicic Kadic A., Boric M., Vidak M., Ferhatovic L. et al. Changes in epidermal thickness and cutaneous innervation during maturation in longterm diabetes // J. Tissue Viability. 2014. Vol. 23. P 7-12.
43. Feletou M. The Endothelium: Part 1: Multiple Functions of the Endothelial Cells – Focus on Endothelium-Derived Vasoactive. San Rafael, CA : Mediators Morgan and Claypool Life Sciences, 2011.
44. Chhabra N. Endothelial dysfunction – a predictor of atherosclerosis // Internet J. Med. Update. 2009. Vol. 4, N 1. P 33-41.
45. Strijdom H., Lochner A. Cardiac endothelium: more than just a barrier! // SA Heart. 2009. Vol. 6, N 3. P 174-185.
46. Mudau M., Genis A., Lochner A., Strijdom H. Endothelial dysfunction: the early predictor of atherosclerosis // Cardiovasc. J. Afr. 2012. Vol. 23, N 4. P. 222-231.
47. Strijdom H. Endothelial dysfunction: are we ready to heed the vasculature’s early-warning signal? // Cardiovasc J. Afr. 2012. Vol. 23, N 4. P. 184-185.
48. Sena C.M., Pereira A.M., Seiga R. Endothelial dysfunction – a major mediator of diabetic vascular disease // Biochim. Biophys. Acta.
2013. Vol. 1832, N 12. P. 2216-2231.
49. Park K.H., Park W.J. Endothelial dysfunction: clinical implications in cardiovascular disease and therapeutic approaches // J. Korean Med. Sci. 2015. Vol. 30, N 9. P 1213-1225.
50. Widmer R.J., Lerman A. Endothelial dysfunction and cardiovascular disease // Glob. Cardiol. Sci. Pract. 2014. Vol. 3. P 291-308.
51. Pushpakumar S., Kundu S., Sen U. Endothelial dysfunction: the link between homocysteine and hydrogen sulfide // Curr. Med. Chem.
2014. Vol. 21, N 32. P. 3662-3672.
52. Tyagi N., Sedoris K.C., Steed M., Ovechkin A.V. et al. Mechanisms of homocysteine-induced oxidative stress // Am. J. Physiol. Heart Circ. Physiol. 2005. Vol. 289, N 6. P. 2649-2656.
53. Woo K.S., Chook P., Lolin Y.I., Cheung A.S. et al. Hyperhomocyst(e) inemia is a risk factor for arterial endothelial dysfunction in humans // Circulation. 1997. Vol. 96. P 2542-2544.
54. Tawakol A., Omland T., Gerhard M., Wu J.T. et al. Hyperhomocyst(e) inemia is associated with impaired endothelium-dependent vasodilation in humans // Circulation. 1997. Vol. 95. P 1119-1121.
55. Bellamy M.F., McDowell I.F., Ramsey M.W., Brownlee M. et al. Hyper-homocysteinemia after an oral methionine load acutely impairs endothelial function in healthy adults // Circulation. 1998. Vol. 98. P 1848-1852.
56. Chambers J.C, Obeid O.A., Kooner J.S. Physiological increments in plasma homocysteine induce vascular endothelial dysfunction in normal human subjects // Arterioscler. Thromb. Vasc. Biol. 1999. Vol. 19. P 29222927.
References
1. Global report on diabetes of WHO. 2016.
2. Tesfaye S., Boulton A.J., Dickenson A.H. Mechanisms and management of diabetic painful distal symmetrical polyneuropathy. Diabetes Care. 2013; 36: 2456-65.
3. The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of Diabetes on the development and progression of long-term complication in insulin-dependent diabetes mellitus. N Engl J Med. 1993; 329: 977-86.
4. Maser R.E., Steenkiste A.R., Dorman J.S., et al. Epidemiological correlates of diabetic neuropathy: report from Pittsburgh Epidemiology of Diabetes Complications Study. Diabetes. 1989; 38 (11): 1456.
5. Wang H., Cui K., Xu K., Xu S. Association between plasma homocysteine and progression of early nephropathy in type 2 diabetic patients. Int J Clin Exp Med. 2015; 8 (7): 11 174-80.
6. Li J., Shi M., Zhang H., Yan L., et al. Relation of homocysteine to early nephropathy in patients with type 2 diabetes. Clin Nephrol. 2012; 77 (4): 305-10.
7. Brazionis L., Rowley K., Itsiopoulos C., Harper C.A., et al. Homocysteine and Diabetic Retinopathy. Diabetes Care. 2008; 31 (1): 50-6.
8. Rudy A., Kowalska I., Str^czkowski M., Kinalska I. Homocysteine concentrations and vascular complications in patients with type 2 diabetes. Diabetes Metab. 2005; 31 (2): 112-7.
9. Kichigina O.N., Golubeva T.I., Troshina I.A., Romanova N.V. Homo-cysteinemia pathogenic role in patients with NAFLD. Meditsinskaya nauka i obrazovanie Urala [Medical Science and Education of the Urals]. 2015; (3): 179. (in Russian)
10. Obeid R., Herrmann W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006; 580 (13): 2994-3005.
11. Poddar R., Paul S. Novel crosstalk between ERK MAPK and p38 MAPK leads to homocysteine-NMDA receptor mediated neuronal cell death. J Neurochem. 2013; 124 (4): 558-70.
12. Skovierova H., Vidomanova E., Mahmood S., Sopkova J., et al. The molecular and cellular effect of homocysteine metabolism imbalance on human health. Int J Mol Sci. 2016; 17 (10): e1733.
13. Cao Y., Chai J.G., Chen Y.C., Zhao J., et al. Beneficial effects of danshensu, an active component of salvia miltiorrhiza, on homocysteine metabolism via the trans-sulphuration pathway in rats. Br J Pharmacol. 2009; 157: 482-90.
14. Selhub J. Homocysteine metabolism. Ann Rev Nutr. 1999; 19: 217-46.
15. Boldyrev A.A. Why is homocysteine toxic? Priroda [Nature]. 2009; (10): 18-23. (in Russian)
16. Perta-Kajan J., Jakubowski H. Paraoxonase 1 and homocysteine metabolism. Amino Acids. 2012; 43: 1405-17.
17. England J.D., Gronseth G.S., Franklin G., Carter G.T., et al.; American Academy of Neurology. Practice parameter: evaluation of distal symmetric polyneuropathy: role of laboratory and genetic testing (an evidence-based review). Report of the American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, and American Academy of Physical Medicine and Rehabilitation. Neurology. 2009; 72: 177-84.
18. Ting R.Z., Szeto C.C., Chan M.H., Ma K.K., et al. Risk factors of vitamin B12 deficiency in patients receiving metformin. Arch Intern Med. 2006; 166: 1975-9.
19. Aroda V.R., Edelstein S.L., Goldberg R.B., et al.; Diabetes Prevention Program Research Group. Long-term metformin use and vitamin B12 deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab. 2016; 101: 1754-61.
20. Berchtold P., Bolli P., Arbenz U., Keiser G. Disturbance of intestinal absorption following metformin therapy (observations on the mode of action of biguanides). Diabetologia. 1969; 5: 405-12.
21. Buvat D.R. Use of metformin is a cause of vitamin B12 deficiency. Am Fam Physician. 2004; 69: 264.
22. American Diabetes Association. 8. Pharmacologic approaches to glycemic treatment. Diabetes Care. 2017; 40 (1): 64-74.
23. Turner L.W., Nartey D., Stafford R.S., Singh S., et al. Ambulatory treatment of type 2 diabetes in the U.S., 1997-2012. Diabetes Care. 2014; 37 (4): 985-92.
24. Nathan D.M., Buse J.B., Davidson M.B., Heine R.J., et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2006; 29: 1963-72.
25. Zalaket J., Wehbe T., Jaoude E. Vitamin B12 deficiency in diabetic subjects taking metformin: a cross sectional study in a Lebanese cohort. J Nutr Intermediary Metab. 2018; 11: 9-13.
26. Wile D.J., Toth C. Association of metformin, elevated homocysteine, and methylmalonic acid levels and clinically worsened diabetic peripheral neuropathy. Diabetes Care. 2010; 33 (1): 156-61.
27. Roy R.P., Ghosh K., Ghosh M., Acharyya A., et al. Study of vitamin B 12 deficiency and peripheral neuropathy in metformin-treated early type 2 diabetes mellitus. Indian J Endocr Metab. 2016; 20: 631-7.
28. Sahin M., Tutuncu N.B., Ertugrul D., Tanaci N., et al. Effects of metformin or rosiglitazone on serum concentrations of homocysteine, folate, and vitamin B12 in patients with type 2 diabetes mellitus. J Diabetes Complications. 2007; 21: 118-23.
29. Pop-Busui R., Sima A., Stevens M. Diabetic neuropathy and oxidative stress. Diabetes Metab Res Rev. 2006; 22 (4): 257-73.
30. Ametov A.S., Kamynina L.L., Rozhdestvenskaya O.A., Pashkova E.Yu. The positive metabolic and antioxidative properties of the testosterone replacement therapy at the combination of the type 2 diabetes mellitus and the hypogonadism. Endokrinologiya: novosti, mneniya, obuchenie [Endocrinology: News, Opinions, Training]. 2016; (3): 83-93. (in Russian)
31. Jacobsen D.W. Hyperhomocysteinemia and oxidative stress. Arte-rioscler Thromb Vasc Biol. 2000; 20: 1182-4.
32. Weiss N., Zhang Y.Y., Heydrick S., Bierl C., et al. Overexpression of cellular glutathione peroxidase rescues homocyst(e)ine-induced endothelial dysfunction. Proc Natl Acad Sci USA. 2001; 98 (22): 12 503-8.
33. Creager M.A., Luscher T.F., Cosentino F., Beckman J.A. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: part I. Circulation. 2003; 108: 1527-32.
34. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001; 414 (6865): 813-20.
35. Malik R.A., Tesfaye S., Thompson S.D., Veves A., et al. Endoneu-rial localisation of microvascular damage in human diabetic neuropathy. Diabetologia. 1993; 36: 454-9.
36. Malik R.A., Newrick P.G., Sharma A.K., Jennings A., et al. Microangiopathy in human diabetic neuropathy: relationship between capillary abnormalities and the severity of neuropathy. Diabetologia. 1989; 32 (2): 92-102.
37. Cameron N.E., Eaton S.E., Cotter M.A., Tesfaye S. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia. 2001; 44: 1973-88.
38. Tuck R.R., Schmelzer J.D., Low P.A. Endoneurial blood flow and oxygen tension in the sciatic nerves of rats with experimental diabetic neuropathy. Brain. 1984; 107 (3): 935-50.
39. Yagihashi S., Mizukami H., Sugimoto K. Mechanism of diabetic neuropathy: where are we now and where to go? J Diabetes Investig. 2011; 2 (1): 18-32.
40. Nedosugova L.V. Diabetic polyneuropathy and oxidative stress. Pathogenesis, diagnosis, treatment: tutorial. Moscow; 2015: 54-5. (in Russian)
41. Schreiber A.K., Nones C., Reis R.C., Chichorro J.G., et al. Diabetic neuropathic pain: physiopathology and treatment. World J Diabetes. 2015; 6 (3): 432-44.
42. Jelicic Kadic A., Boric M., Vidak M., Ferhatovic L., et al. Changes in epidermal thickness and cutaneous innervation during maturation in long-term diabetes. J Tissue Viability. 2014; 23: 7-12.
43. Feletou M. The Endothelium: Part 1: multiple functions of the endothelial cells focus on endothelium-derived vasoactive. San Rafael, CA: Mediators Morgan and Claypool Life Sciences, 2011.
44. Chhabra N. Endothelial dysfunction – a predictor of atherosclerosis. Internet J Med Update. 2009; 4 (1): 33-41.
45. Strijdom H., Lochner A. Cardiac endothelium: more than just a barrier! SA Heart. 2009; 6 (3): 174-85.
46. Mudau M., Genis A., Lochner A., Strijdom H. Endothelial dysfunction: the early predictor of atherosclerosis. Cardiovasc J Afr. 2012; 23 (4): 222-31.
47. Strijdom H. Endothelial dysfunction: are we ready to heed the vasculature’s early-warning signal? Cardiovasc J Afr. 2012; 23 (4): 184-5.
48. Sena C.M., Pereira A.M., Seiga R. Endothelial dysfunction a major mediator of diabetic vascular disease. Biochim Biophys Acta. 2013; 1832 (12): 2216-31.
49. Park K.H., Park W.J. Endothelial dysfunction: clinical implications in cardiovascular disease and therapeutic approaches. J Korean Med Sci. 2015; 30 (9): 1213-25.
50. Widmer R.J., Lerman A. Endothelial dysfunction and cardiovascular disease. Glob Cardiol Sci Pract. 2014; 3: 291-308.
51. Pushpakumar S., Kundu S., Sen U. Endothelial dysfunction: the link between homocysteine and hydrogen sulfide. Curr Med Chem. 2014; 21 (32): 3662-72.
52. Tyagi N., Sedoris K.C., Steed M., Ovechkin A.V., et al. Mechanisms of homocysteine-induced oxidative stress. Am J Physiol Heart Circ Physiol. 2005; 289 (6): 2649-56.
53. Woo K.S., Chook P., Lolin Y.I., Cheung A.S., et al. Hyperhomocyst(e) inemia is a risk factor for arterial endothelial dysfunction in humans. Circulation. 1997; 96: 2542-4.
54. Tawakol A., Omland T., Gerhard M., Wu J.T., et al. Hyperhomocyst(e) inemia is associated with impaired endothelium-dependent vasodilation in humans. Circulation. 1997; 95: 1119-21.
55. Bellamy M.F., McDowell I.F., Ramsey M.W., Brownlee M., et al. Hyperhomocysteinemia after an oral methionine load acutely impairs endothelial function in healthy adults. Circulation. 1998; 98: 1848-52.
56. Chambers J.C, Obeid O.A., Kooner J.S. Physiological increments in plasma homocysteine induce vascular endothelial dysfunction in normal human subjects. Arterioscler Thromb Vasc Biol. 1999; 19: 2922-7.