 Пандемия ожирения и сопутствующих состояний неалкогольной жировой болезни печени , атеросклероза и диабета 2 типа (СД2) поднимает проблему резистетности к инсулину. Связь между ожирением, инсулинорезистентностью и T2D давно известна. Резистентность к инсулину является общей чертой всех этих заболеваний, и было приложено немало усилий для определения патогенеза резистентности к инсулину.
Пандемия ожирения и сопутствующих состояний неалкогольной жировой болезни печени , атеросклероза и диабета 2 типа (СД2) поднимает проблему резистетности к инсулину. Связь между ожирением, инсулинорезистентностью и T2D давно известна. Резистентность к инсулину является общей чертой всех этих заболеваний, и было приложено немало усилий для определения патогенеза резистентности к инсулину.
Интересна роль инсулина и питательных веществ (в частности, глюкозы и жирных кислот) в хранении питательных веществ , а также различных дефектов этого процесса , которые вызывают резистентность к инсулину и сахарный диабет 2 типа. Раннее предполагалось, что резистентность мышечного инсулина к ожирению можно объяснить повышенным окислением жирных кислот, что ограничивает стимулированное инсулином использование глюкозы. Основным принципом этой модели было то, что увеличение доставки и окисления жирных кислот приводит к накоплению цитрата, который ингибирует фосфофруктокиназу, ключевой фермент в гликолизе, что приводит к увеличению внутриклеточного глюкозо-6-фосфата и глюкозы, что тем самым ухудшает утилизацию глюкозы.
Простой акт приема пищи быстро смещает метаболизм глюкозы в печени из производства глюкозы в накопление глюкозы, что представляет собой сложный переход, регулируемый множеством факторов, включая питательные вещества, изменения в гормонах поджелудочной железы и кишечника и регуляцию со стороны нервной системы. Инсулин является важнейшим регулятором этого перехода, прежде всего, путем активации гликогенсинтазы. Значение инсулина наблюдается у пациентов с диабетом 1 типа (СД1), которые синтезируют только одну треть необходимого количества печеночного гликогена после смешанного приема пищи. Тем не менее, гиперинсулинемия, при отсутствии гипергликемии, способствует образованию циклического гликогена в печени ( с минимальным чистым синтезом гликогена в печени). А гипергликемия без гиперинсулинемии ингибирует гликогенолиз в печени через глюкозо-опосредованное ингибирование гликогенфосфорилазы. Сочетание гиперинсулинемии и гипергликемии максимизирует чистый синтез гликогена в печени. Другие питательные вещества дополнительно оптимизируют чистый синтез гликогена в печени, такой как активация глюкокиназы каталитическими количествами фруктозы.
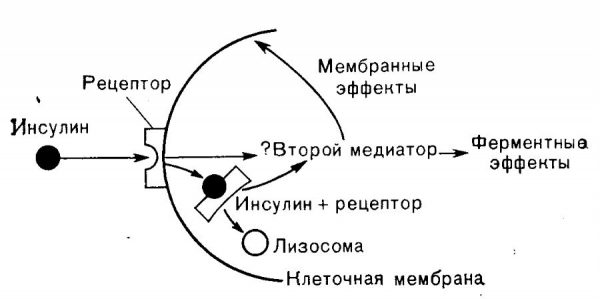
Действие печеночного инсулина требует скоординированной передачи внутриклеточных сигналов. Инсулин активирует инсулиновую рецепторную тирозинкиназу (IRTK) с последующей активацией киназ, включая 3-фосфоинозитид-зависимую киназу-1 (PDK1) и mTORC2 , которые сходятся при фосфорилировании Akt. Характер доставки инсулина также может влиять на фосфорилирование Akt, при этом пульсирующая портальная доставка (которая лучше имитирует физиологию) приводит к большей активации, чем непрерывная фиксированная доставка инсулина. Активация Akt является неотъемлемым результатом многочисленных процессов необходимых для регуляции глюкозы и липидного обмена в печени. Эта модель использовалась для объяснения того, как инсулин подавляет выработку глюкозы в печени через (i) снижение экспрессии глюконеогенных ферментов через фосфорилирование и ядерное исключение FOXO1 и (ii) инактивацию гликогенсинтазинкиназы 3β (GSK3β), которая позволяет активировать гликогенсинтазу.
Последние исследования ставят под сомнение примат Akt / GSK3β / гликогенсинтазной ветви в регуляции синтеза гликогена. Таким образом, активация Akt не является стержнем для координации метаболизма глюкозы в печени, и другие пути также регулируют этот процесс. Опосредованное инсулином подавление выработки глюкозы в печени часто объясняется снижением транскрипции глюконеогенных ферментов; однако эта модель не может объяснить быстрое подавление выработки глюкозы в печени после еды.
Липиды являются вторым основным источником топлива. Пищевые липиды преимущественно попадают в системный кровоток в виде аполипопротеинов B48 (содержащих ApoB48) триглицерид-богатых хиломикронов, которые быстро очищаются. Гидролиз триглицеридов в жирные кислоты с помощью липопротеинлипазы (LpL) является регулирующим этапом для поглощения клеточных липидов из плазмы и строго регулируется. Высвобождение триглицеридов из внутриклеточных хранилищ также точно контролируется внутриклеточными липазами. Инсулин увеличивает экспрессию LpL в подкожно-жировой клетчатке , в то время как резистентность к инсулину у людей связана с уменьшением экспрессии подкожно-жировой клетчатки. В то время как инсулин регулирует базальную экспрессию LpL, ряд других белков регулирует активность LpL.
Терапия лептином позволяет снизить потребление калорий, устранить стеатоз печени и улучшить действие инсулина. Кратковременные диеты с высоким содержанием жиров приводят к печеночному стеатозу и печеночной резистентности к инсулину без накопления мышечного липида или периферической резистентности к инсулину. Есть некоторые обстоятельства, при которых печеночный стеатоз кажется диссоциированным от печеночной резистентности к инсулину. Снижение экспрессии сравнительной идентификации гена-58 (CGI-58), активатора жировой триглицеридной липазы (ATGL), привело к выраженному стеатозу печени и увеличению общего содержания DAG без резистентности к печеночному инсулину.
Частично, устойчивость к жировому инсулину – это неспособность инсулина активировать транспорт жировой глюкозы, стимулировать поглощение липидов и подавлять липолиз. Хотя снижение поглощения глюкозы в жировой ткани продемонстрировано как на моделях in vivo, так и in vitro, механизм и метаболическое влияние нарушения инсулино-опосредованного поглощения глюкозы в жировой ткани остаются неясными. Стимулируемый инсулином метаболизм глюкозы в жировой ткани количественно незначителен по сравнению с метаболизмом глюкозы, стимулированным инсулином в целом организме; однако метаболизм глюкозы в адипоцитах оказывает экстра-жировое действие. Например, потеря жирового GLUT4 у мышей не изменяет ожирение или увеличение веса, но приводит к резистентности к инсулину в скелетных мышцах и печени.
Адипоциты выделяют специфические жирные кислоты, которые связаны с повышенной чувствительностью к инсулину, такие как пальмитолеат или монометиловые жирные кислоты с разветвленной цепью. Адипоцитокины (например, лептин, резистин и адипонектин) также влияют на системную чувствительность к инсулину. Увеличение лептина и резистина связано с резистентностью к инсулину, в то время как адипонектин связан с повышенной чувствительностью к инсулину.
Центральным парадоксом в патогенезе СД2 является очевидная селективная природа резистентности к инсулину в печени, при которой инсулин не способен подавлять выработку глюкозы в печени, но продолжает стимулировать липогенез, что приводит к гипергликемии, гиперлипидемии и стеатозу печени. Усилия по объяснению этого парадокса были сосредоточены на поиске точки ветвления в передаче сигналов инсулина, где печеночная глюкоза и липидный обмен расходятся. Было выссказано предположил, что синтез триглицеридов в печени может быть вызван субстратом (жирными кислотами), независимо от изменений в передаче сигналов инсулина в печени.
Моносахариды действуют как субстраты и регуляторы питательных веществ печеночных DNL. У нормальных субъектов глюкоза в основном метаболизируется в скелетных мышцах. Тем не менее, мышечная резистентность к инсулину, возникающая на ранних стадиях развития СД2 , увеличивает доставку глюкозы в печень, где липогенный механизм вызван хронической гиперинсулинемией. Фруктоза, которая потребляется почти в таких же количествах, что и глюкоза, также может способствовать липогенезу. Как глюкоза, так и фруктоза также могут регулировать печеночный липогенез независимо от действия инсулина.
Дисбаланс калорий, поддерживаемый нашей современной токсичной средой, приводит к эктопическому накоплению липидов в печени и скелетных мышцах, что препятствует действию инсулина в этих тканях. Последующая дисфункция адипоцитов способствует инфильтрации макрофагов и увеличивает липолиз, еще более нарушая метаболизм углеводов и липидов в печени несколькими путями. Увеличение потока жирных кислот способствует этерификации жирных кислот и синтезу триглицеридов в печени, усугубляя стеатоз печени, резистентность к инсулину в печени и гипертриглицеридемию.