Резюме
В обзорной статье представлен анализ клинических и экспериментальных данных о патогенетических механизмах инсулинорезистентости и липотоксичности при сахарном диабете. Рассматриваются их взаимосвязь и роль в развитии нарушения углеводного обмена с позиции теории ограниченной расширяемости жировой ткани. Также проанализированы протективные свойства адипонектина за счет повышения чувствительности к инсулину, стимуляции окисления свободных жирных кислот в печени и мышцах. Фармакотерапия инсулинорезистентности и липотоксичности в настоящее время ограничена. Отечественный гипогликемический комплексный препарат, содержащий антитела к С-концевому фрагменту β-субъединицы рецептора инсулина и антитела к эндотелиальной NO-синтазе в релиз-активной форме, показал эффективность в отношении чувствительности тканей к инсулину и повышения продукции адипонектина, что делает возможным его использование в качестве дополнительного компонента сахароснижающей терапии.
Ключевые слова:ожирение, сахарный диабет типа 2, инсулинорезистентность, липотоксичность, свободные жирные кислоты, адипонектин
Для цитирования: Аметов А.С., Тертычная Е.А. Инсулинорезистентность и липотоксичность – две грани одной проблемы при сахарном диабете типа 2 и ожирении // Эндокринология: новости, мнения, обучение. 2019. Т. 8, № 2. С. 25-33. doi: 10.24411/2304-9529-2019-12003.
Ожирение и сахарный диабет типа 2 (СД2) в последние десятилетия стали важнейшими медицинскими, социальными и экономическими проблемами. По данным Федерального регистра СД, в 2017 г. количество пациентов с СД2 составило 4 млн [1]. По сообщению Минздрава России, в 2017 г. ожирение впервые выявлено у 1,9 млн россиян. Распространенность ожирения среди мужчин в возрасте от 25 до 64 лет составляет 26,9%, среди женщин – 30,8% [2]. Неинфекционные эпидемии стали проблемой № 1 здравоохранения развитых стран и требуют решительных, безотлагательных мер. Согласно современным представлениям, модификация образа жизни в комплексе с ранней комбинированной терапией позволит избежать дальнейшего развития СД2 и его осложнений.
Теория ограниченной расширяемости жировой ткани
Жировая ткань (ЖТ) обладает высокой метаболической активностью, осуществляет синтез и секрецию множества биологически активных веществ, участвующих в углеводном (адипонектин, резистин, висфатин, оментин) и жировом обмене (белок – переносчик холестериновых эстераз, перилипин, ретинол-связывающий белок, аполипопротеин Е), процессах воспаления [фактор некроза опухоли α (ФНОα), интерлейкины (ИЛ-6, ИЛ-8, ИЛ-10, ИЛ-12), С-реактивный белок, адипсин, апелин, резистин], свертывания крови [ингибитор активатора плазминогена-1 (ИАП-1), тканевый фактор], поддержания артериального давления (ангиотензин II, ангиотензиноген, апелин) и пищевого поведения (лептин) [3]. Выделяют абдоминальный и висцеральный типы жировых отложений, между которыми существуют значительные функциональные различия. Висцеральный жир, расположенный около внутренних органов, брыжейки и сальника, отличается от подкожного типом адипоцитов, их эндокринной функцией, липолитической активностью, чувствительностью к инсулину и другим гормонам. Например, в висцеральной жировой ткани выше экспрессия и секреция ИЛ-6, ИАП-1, адипонектина, ангиотензиновых рецепторов 1-го типа, β3-адренергических, глюкокортикоидных и андрогенных рецепторов, а лептина – в подкожной. Более того, адипокины висцеральной жировой ткани поступают преимущественно в портальную систему и в печень, тогда как из подкожных депо – непосредственно в системный кровоток [4].
Отличительной чертой ожирения с висцеральным типом отложений является нарушение процесса подавления высвобождения свободных жирных кислот (СЖК) в ответ на инсулин и прием пищи по сравнению с пациентами без невыраженного ожирения или с ним [5, 6]. Уровень СЖК в постпрандиальном периоде у пациентов с висцеральным ожирением в 3 раза выше, что доказывает повышенную резистентность жировых клеток к антилиполитическому эффекту инсулина [6]. В исследованиях in vitro хроническое повышение СЖК (преимущественно пальмитиновой, линоленовой и стеариновой кислот) приводило к подавлению глюкозостимулированной секреции инсулина β-клетками с последующим их апоптозом, а также к развитию инсулинорезистентности в печени и мышцах [7]. Данный комплекс патологических событий получил название “липотоксичность”. Клиническими исследованиями подтверждается взаимосвязь липотоксичности и СД2. В исследовании M. Korani и соавт. общий уровень насыщенных и мононенасыщенных жирных кислот у пациентов с СД был статистически значимо выше, чем в контрольной группе (р=0,006, 0,02 соответственно). Кроме того, у пациентов с СД2 уровень полиненасыщенных ЖК в сыворотке был значительно ниже, чем у здоровых людей (р=0,02). Заболеваемость СД2 положительно коррелировала с уровнями пальмитиновой, насыщенных и мононенасыщенных жирных кислот [8].
Взаимосвязь высоких концентраций СЖК с резистентностью к инсулину мышечной ткани была показана в исследовании M. Bajaj и соавт. [9]. Пациенты с СД2 получали терапию аципимоксом (ингибитор липолиза) по 250 мг каждые 6 ч в течение 7 дней, в результате было достигнуто значимое снижение уровня СЖК, которое коррелировало с повышением чувствительности к инсулину [9]. Похожие результаты были продемонстрированы на пациентах, имеющих генетическую предрасположенность к развитию СД [10].
На основании дальнейших исследований была сформулирована теория ограниченной расширяемости жировой ткани (рис. 1). В условиях положительного энергетического баланса ЖТ накапливает избытки СЖК в виде триглицеридов (ТГ), однако ее возможности к накоплению ограничены преимущественно генетическими факторами, а также особенностями висцерального жира. В данной ситуации инсулинорезистентность может выполнять защитную функцию для адипоцитов от избыточного накопления СЖК, которые поступают с пищей. В результате в инсулинорезистентных, “переполненных” ТГ адипоцитах активируется процесс липолиза, СЖК покидают ЖТ и попадают в систему портальной вены, оттуда поступая в печень и далее в другие органы и ткани [11].
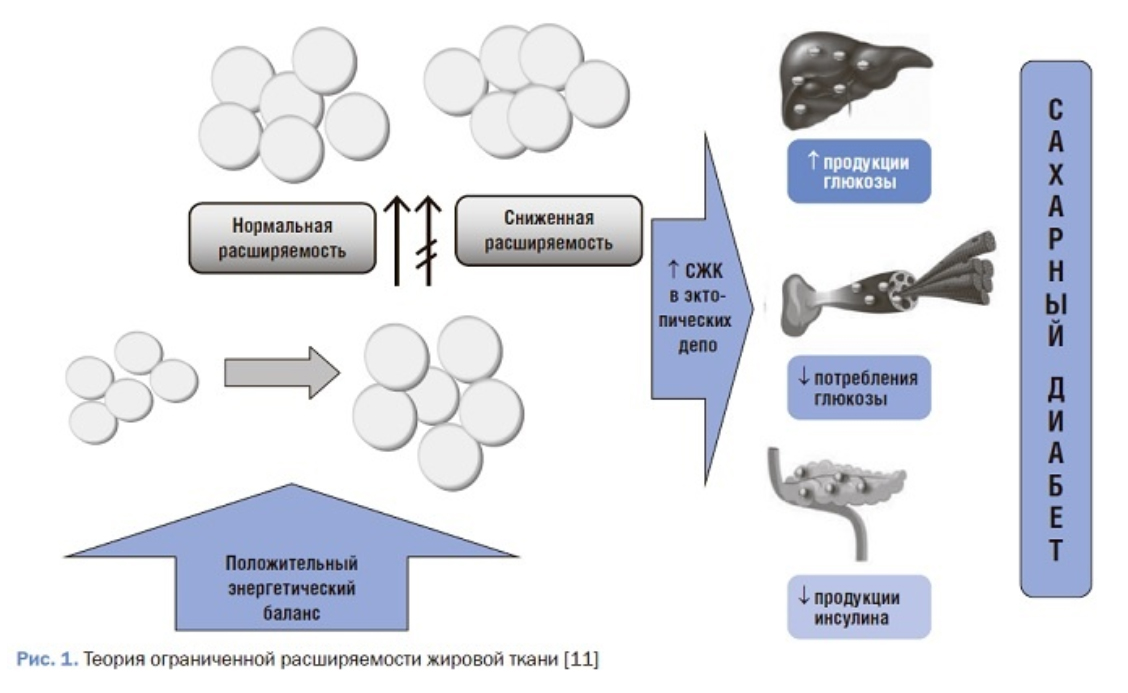
Биохимические процессы, которые нарушаются под воздействием СЖК, сходны в различных органах тканях. В настоящее время получены данные об их патологических эффектах в р-клетках поджелудочной железы, печени, мышцах, сердце, почках.
Роль церамида и диацилглицерола в развитии инсудинорезистентности и липотоксичности
Церамиды – это мембранные липиды, предшественники сфингомиелина [12]. В 1990 г. впервые было доказано, что увеличение содержания церамида в печени и мышцах наряду с диацилглицеролами связано с резистентностью к инсулину у крыс (fa/fa крыс, гомозиготных по усеченному нефункциональному рецептору лептина) [13]. Воздействие избыточных длинноцепочечных насыщенных СЖК [пальмитата (16:0), стеарата (C18:0), арахидата (C20:0)] и линоцерата (C24:0), но не более короткого [миристата (C12:0)] или ненасыщенных СЖК индуцирует накопление церамидов с помощью серин-пальмитоилтрансферазы и церамидсин-тазы [14]. Предположительно синтез de novo церамида является медиатором СЖК-индуцированной гибели β-клеток. Сверхэкспрессия церамидсинтазы потенцирует пальмитат-индуцированное накопление церамидов и усиливает апоптоз путем продуцирования токсичных видов церамидов, таких как C18:0, C22:0 и C24:1 [15]. С2-церамид способен потенцировать проапоптоз и антипролиферативный эффект пальмитиновой кислоты в р-клетках. ЭР-стресс, изменения целостности митохондриальной мембраны и ингибирование протеинкиназы В церамидом предположительно индуцируют апоптоз в β-клетках. В исследовании E. BosLem и соавт. было доказано, что при введении в р-клетки синтетического церамида происходит его накопление в ЭР, снижаются уровни сфингомиелина и холестерина, что приводит к нарушению липидных плотов [16]. Церамид увеличивает проницаемость митохондриальной мембраны и приводит к активации внутренних путей через антиапоптотические молекулы BcL-2 и увеличение каспазы 3/7 [15]. Инактивация протеинкиназы В ассоциирована с накоплением церамидов и наблюдается у человека, получающего насыщенный жир. Ингибирование синтеза de novo церамида ослабляет СЖК-индуцированный р-клеточный апоптоз и снижает гипергликемию [17].
Роль мембранных белков-переносчиков в развитии инсулинорезистентности и липотоксичности
СЖК поступают внутрь клеток путем пассивной диффузии и с помощью транспортных белков: транслоказы жирных кислот (CD36), белка, связанного с плазматической мембраной, связывающей жирные кислоты (FABPpm), и белка транспорта жирных кислот (FATP). CD36 – ключевой переносчик ЖК в сердце и скелетных мышцах. Он находится на плазматической и митохондриальной мембране. Основным регулятором экспрессии данного белка является инсулин через путь передачи сигналов протеинкиназы B. При гиперинсулинемии увеличивается цитозольная экспрессия CD36, а также сопутствующее повышение содержания митохондриального CD36 в мышечных клетках. Таким образом, в митохондриях усиливается окисление ЖК и, как следствие, синтез из них церамидов. В β-клетках CD36 выполняет аналогичные функции. При гипергликемии увеличивается экспрессия данного белка на мембране, в результате поток ЖК в клетку приводит к активации оксидативного стресса и провоспалительных цитокинов. Учитывая важную роль данного транспортного белка, были созданы селективные ингибиторы CD36 – сульфо-N-сукцинимидильные производные. Предварительная инкубация с ингибитором CD36 предотвращала СЖК-индуцированный апоптоз с помощью продукции восстановленного активного кислорода.
Другой предполагаемый переносчик жирных кислот -FABPpm – увеличивает скорость транспорта СЖК через сарколемму, однако не участвует в их транспорте в митохондрии. FATP представляют собой семейство из 6 интегральных мембранных белков с внеклеточным/просветным N- и C-концевым доменами с активностью жирной ацил-КоА-синтетазы; следовательно, белки FATP обладают способностью захватывать жирные кислоты внутриклеточно. Изучение данных белков в настоящее время продолжается [18].
Роль митохондрий в развитии инсулинорезистентности и липотоксичности
Митохондрии являются основным местом деградации липидов. Клетки защищают себя от липотоксичности и гибели, либо окисляя СЖК, либо изолируя их в ТГ в виде липидных капель (ЛК). ТГ синтезируются с помощью ацилтрансфераз и фосфатаз в саркоплазматической сети и митохондриальной мембране, а затем упаковываются в цитоплазматические ЛК [19]. Чрезмерное накопление ЛК в гепатоцитах, кардиомиоцитах, скелетных миоцитах, адренокортикальных клетках, энтероцитах и макрофагах является отличительной чертой СД2, ожирения, атеросклероза, стеатоза печени и других метаболических заболеваний [20]. Хорошо известно, что митохондриальная функция необходима для нормальной стимулированной глюкозой секреции инсулина из β-клеток поджелудочной железы [21]. K. Peterson и соавт. в 2003 г. показали, что снижение синтеза аденозинтрифосфата (АТФ) связано с увеличением внутриклеточного жира. В изолированных митохондриях от людей, страдающих ожирением и СД2, синтез АТФ снижается и тесно коррелирует с уменьшением инсулин-стимулированной утилизации глюкозы и увеличением СЖК в плазме натощак [22]. Окислительная активность митохондрий и синтез АТФ снижаются, эти данные подтверждают гипотезу о том, что резистентность к инсулину у человека возникает из-за дефектов окисления митохондриальных жирных кислот, которые, в свою очередь, приводят к увеличению внутриклеточных метаболитов жиров. Предполагается, что снижение митохондриальной окислительно-фосфорилирующей активности у лиц с инсулинорезистентностью связано с нарушением функции митохондрий [12].
Роль эндоплазматического ретикулума в развитии инсулинорезистентности и липотоксичности
Активация стресса эндоплазматического ретикулума (ЭР) играет важную роль в этиологии резистентности к инсулину, особенно наблюдаемой при неалкогольной жировой болезни печени [12]. Сниженная способность ЭР к сворачиванию белка вызывает накопление незрелых белков в просвете ЭР, и это запускает сложную, в первую очередь адаптивную сигнальную сеть, называемую ответом развернутого белка или стрессом ЭР. Данный механизм регулирует липогенез, позволяя расширять мембрану ЭР и увеличивая его способность обрабатывать белки [12]. Когда стресс ЭР не может восстановить его функции, апоптоз стимулируется в основном за счет индукции гомологичного белка, связывающего белок CCAAT/энхансер (CHOP) и активации JNK. Активация JNK также запускается окислительным стрессом при липотоксичности [23]. Помимо своей проапоптотической активности, JNK также препятствует передаче сигналов инсулина путем фосфорилирования субстрата-1 рецептора инсулина в серин, который представляет собой ключевую связь между ЭР-стрессом и инсулинорезистентностью [24]. Способность стресса в ЭР вызывать печеночную инсулинорезистентность может в конечном итоге зависеть от того, изменяется ли баланс липогенеза и экспорта липидов. Запрограммированная гибель клеток, вызванная СЖК, которая, по крайней мере частично, происходит из-за стресса ЭР, называется липоапоптозом.
В ЖТ активация стресса ЭР, по-видимому, регулирует энергетический баланс. Мыши с гетерозиготной делецией Grp78 имели активацию адаптивных факторов стресса ЭР и были защищены от ожирения, стеатоза печени и резистентности к инсулину [25]. Данные свидетельствуют о том, что стресс ЭР функционирует как часть клеточного ответа, чтобы сбалансировать метаболические потребности. Некоторые аспекты стресса ЭР четко регулируют липогенез (например, путь IRE1a-XBP1s), образование ЛК и накопление липидов (например, через АТФ), а также регулируют метаболизм глюкозы [26]. Таким образом, активация стресса ЭР может изменять клеточный липидный баланс и нарушать передачу сигналов инсулина [12].
Помимо указанных патологических механизмов, в развитии инсулинорезистентности и липотоксичности также участвует активация провоспалительных цитокинов, оксидативного стресса [27]. Увеличение продукции АФК приводит к повреждению ДНК, белков и липидов. Он активирует определенные чувствительные к стрессу сигнальные пути, такие как ядерный фактор κB, p38MAPK и JNK [28], которые в конечном счете способствуют гибели клеток. Повышенный окислительный стресс также влияет на окислительно-восстановительный гомеостаз в ЭР [29].
Активация сигнального пути протеинкиназы С
Протеинкиназа С (ПКС) является ферментом, который связан с резистентностью к инсулину в печени. Было показано, что ПКС активируется в печени крыс, подвергнутых 6-часовому введению интралипида/гепарина, наряду с развитием резистентности к печеночному инсулину [30]. Эффекты ПКС могут быть опосредованы через измененный липогенез; экспрессия ключевых липогенных ферментов снижается у нокаутных по ПКС мышей. Эти данные могут служить доказательством того, что опосредованная диацилглицеролом активация ПКС напрямую нарушает передачу сигналов и действие инсулина, объясняя резистентность к инсулину как в мышцах, так и в печени при избытке липидов.
Более 20 лет изучается взаимосвязь инсулинорезистентности и липотоксичности, накоплено большое количество
теоретических данных об этих патологических процессах, однако человеческий организм имеет и защитные механизмы. Один из них – гормон ЖТ адипонектин.
Протективная роль адипонектина
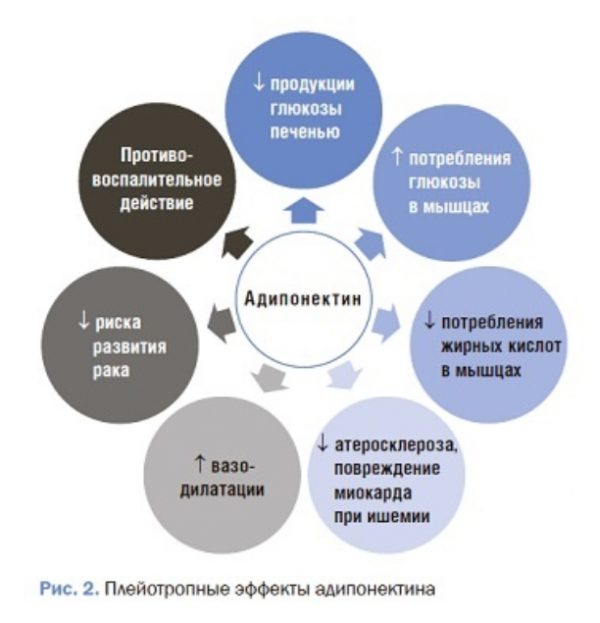 Адипонектин был открыт в 1995 г. Он оказывает плейотропное действие: повышает чувствительность к инсулину, стимулирует окисление СЖК в печени и мышцах, ингибирует апоптоз, антиатерогенное и противовоспалительное действие, снижает образование раковых клеток (рис. 2) [4]. В 2001 г. две ключевые работы впервые продемонстрировали физиологическую роль адипонектина. После ведения адипонектина мышам его концентрация в плазме увеличивалась в 4 раза, что приводило к инсулинонезависимому снижению уровня глюкозы через подавление глюконео-генеза [31]. В другом исследовании T. Yamauchi и соавт. показали, что адипонектин может увеличивать β-окисление в скелетных мышцах и подавлять накопление липидов в печени через активацию 5’AMP-активируемой протеинкиназы (AMPK) [32].
Адипонектин был открыт в 1995 г. Он оказывает плейотропное действие: повышает чувствительность к инсулину, стимулирует окисление СЖК в печени и мышцах, ингибирует апоптоз, антиатерогенное и противовоспалительное действие, снижает образование раковых клеток (рис. 2) [4]. В 2001 г. две ключевые работы впервые продемонстрировали физиологическую роль адипонектина. После ведения адипонектина мышам его концентрация в плазме увеличивалась в 4 раза, что приводило к инсулинонезависимому снижению уровня глюкозы через подавление глюконео-генеза [31]. В другом исследовании T. Yamauchi и соавт. показали, что адипонектин может увеличивать β-окисление в скелетных мышцах и подавлять накопление липидов в печени через активацию 5’AMP-активируемой протеинкиназы (AMPK) [32].
В более поздних исследованиях была доказана роль фермента церамидазы в плейотропных эффектах адипонектина. Церамидаза – это фермент, превращающий вредные церамиды в полезный класс липидов, сфинганинов и сфингозинов, включая подкласс сфингозин-1-фосфата (S1P). Было доказано, что содержание церамидов в печени увеличивается у мышей ob/ob с генетическим ожирением и у животных с ожирением, вызванным высокожировой диетой [33]. Введение адипонектина приводит к сильному снижению уровня печеночных церамидов и, следовательно, к сенсибилизации к инсулину. Кроме того, у животных, получавших высокожировую диету, генетически обусловленное повышение уровня адипонектина связано с улучшенной толерантностью к глюкозе и сниженным содержанием церамида. Недавно
Помимо мощных эффектов на периферии, адипонектин также оказывает центральное действие. Введение адипонектина путем внутривенной инъекции приводит к перемещению белка через гематоэнцефалический барьер и повышает уровень адипонектина в спинномозговой жидкости [35]. В то же время внутрицеребровентрикулярная инъекция адипонектина вызывает снижение массы тела и увеличение расхода энергии, что может быть обусловлено повышением регуляции гипоталамического кортикотропин-рилизинг-гормона. Кроме того, адипонектин был обнаружен в спинномозговой жидкости [36]. Необходимы дальнейшие исследования для изучения сложного распределения и физиологического действия адипонектина в мозге в клинически значимых условиях у человека.
В исследованиях было доказано, что уровень циркулирующего адипонектина повышается при снижении массы тела. Диета, шунтирование желудка, орлистат или римонабант значительно повышали уровни адипонектина параллельно с уменьшением массы тела [37]. Тиазолидиндионы увеличивали экспрессию адипонектина и его уровень в крови у грызунов, лиц без диабета и пациентов с СД2. Упражнения без значительной потери массы тела не влияли на циркулирующий адипонектин, но улучшали чувствительность к инсулину. Экспрессия гена адипонектина и уровень циркулирующего адипонектина ниже у пациентов с СД2 по сравнению со здоровыми людьми [38]. В проспективном исследовании в течение 5 лет наблюдали 3599 мужчин без диабета, и было доказано, что низкий уровень адипонектина связан с повышенным риском развития СД2, даже после корректировки на традиционные факторы риска [39]. Таким образом, адипонектин можно считать в высшей степени перспективной молекулой для коррекции инсулинорезистентности.
Возможности фармакотерапии инсулинорезистентности и липотоксичности
Выше изложено множество сложных и взаимосвязанных биохимических процессов, которые вместе и по отдельности приводят к катастрофическим последствиям в организме. В реальной практике, как правило, пациенты попадают на прием к врачу, когда весь патологический цикл запущен и можно увидеть последствия: развернутую клиническую картину СД2, дислипидемии, атеросклероза и нередко поздних осложнений. В данной ситуации врач должен в доступной форме объяснить принципы рационального питания, физической нагрузки, которые, по данным многочисленных исследований, способны прервать большинство патологических механизмов. Следует особенно подчеркнуть важность участия пациента в своем лечении, так как именно от его мотивации во многом зависит результат. Одним из современных подходов к достижению метаболического контроля является ранняя комбинированная патогенетическая терапия. Данный подход позволяет “не теряя времени” помочь метаболизму вернуться на “правильный путь”, активировать защитные механизмы и предотвратить развитие тяжелых осложнений СД2 [40].
Вне всяких сомнений, наиболее патогенетически выгодной комбинацией являются сочетания, содержащие метформин как основной препарат для улучшения чувствительности тканей к инсулину. Выбор второго препарата осуществляется лечащим врачом в соответствии с индивидуальными целями для каждого пациента, прежде всего в соответствии с сердечно-сосудистой коморбидностью. В данном контексте необходимо обратить внимание на препараты отечественной фармацевтики.
Комплексный препарат, содержащий антитела к С-концевому фрагменту р-субъединицы рецептора инсулина (релиз-активные антитела) и антитела к эндотелиальной NO-синтазе, оказывает плейотропное действие: повышает чувствительность тканей к инсулину, что сопровождается умеренным снижением уровня глюкозы натощак на 1,15 ммоль/л ко 2-й неделе терапии, а также позволяет повысить эффективность инсулинотерапии, стабилизировать применяемые дозы препаратов инсулина и снизить риск развития побочных эффектов [41-43]. Кроме того, препарат стимулирует работу фермента эндотелиальной NO-синтазы и продукции оксида азота, что способствует снижению реактивности сосудов, уменьшению сосудистого спазма, нормализации уровня артериального давления и улучшению периферической микроциркуляции [44, 45]. Особый интерес представляют данные исследования, проведенные на зрелых адипоцитах человека, которые инкубировали в течение 72 ч вместе с комплексным препаратом антител к С-концевому фрагменту β-субъединицы рецептора инсулина и антител к эндотелиальной NO-синтазе [46]. Было показано статистически значимое увеличение концентрации адипонектина в культуральной среде по сравнению с показателями, полученными для контролей. Препарат сравнения росиглитазон стимулировал продукцию адипонектина, но выраженность эффекта была ниже по сравнению с эффектом антител к С-концевому фрагменту β-субъединицы рецептора инсулина и антител к эндотелиальной NO-синтазе. Поскольку культуральная среда не содержала инсулин, полученные данные позволяют предположить, что комбинированный препарат посредством прямого влияния на β-субъединицу рецептора инсулина зрелых адипоцитов человека активирует рецептор инсулина, что вызывает активацию его сигнальных путей, приводя к усилению секреции адипонектина [46]. Учитывая ограниченный выбор препаратов, улучшающих чувствительность тканей к инсулину, комбинация антител к С-концевому фрагменту β-субъединицы рецептора инсулина и антител к эндотелиальной NO-синтазе может быть использована в качестве дополнительного компонента сахароснижающей терапии.
Благодаря многочисленным экспериментальным и клиническим исследованиям не вызывает сомнений тесная взаимосвязь инсулинорезистентности и липотоксичности в патогенезе ожирения и СД2. Поиск и разработка молекул, способных эффективно влиять на данные патологические механизмы, являются одними из важнейших проблем современной эндокринологии. Возможность влиять на уровень адипонектина представляется перспективным способом коррекции как инсулинорезистентности, так и липотоксичности, благодаря его плейотропным эффектам. В то время как создание препаратов на основе данного вещества является вопросом будущего, в отечественной фармацевтике уже существуют молекулы, способные опосредованно влиять на инсулинорезистентность и уровень адипонектина, что расширяет возможности ранней комбинированной терапии СД2.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Литература
1. Дедов И.И., Шестакова М.В., Викулова О.К. Эпидемиология сахарного диабета в Российской Федерации: клинико-статистический анализ по данным Федерального регистра сахарного диабета // Сахарный диабет. 2017. № 20 (1). С. 13-41.
2. Минздрав назвал страдающие от ожирения регионы. https:// www.rbc.ru/society/24/07/2018/5b519ee49a7947f2d4d7fa9b
3. Hajer G.R., wan Haeften T.W., Visseren F.L. Adipose tissue disfunction in obesity, diabetes and vascular diseases // Eur. Heart J. 2008. Vol. 29, N 24. P. 2959-2971. doi: 10.1093/eurheartj/ehn387
4. Дедов И.И., Мельниченко Г.А., Бутрова С.А. Жировая ткань как эндокринный орган // Ожирение и метаболизм. 2006. № 1. P 6-13.
5. Guo Z., Hensrud D.D., Johnson C.M., Jensen M.D. Regional postprandial fatty acid metabolism in different obesity phenotypes // Diabetes. 1999. Vol. 48, N 8. P. 1586-1592.
6. Ebbert J.O., Jensen M.D. Fat depots, free fatty acids, and dyslipidemia // Nutrients. 2013. Vol. 5, N 2. P. 498-508.
7. Unger R.H. Lipotoxic diseases // Ann. Rev. Med. 2002. Vol. 53. P. 319-336.
8. Korani M., Firoozrai M., Maleki J., et al. Fatty acid composition of serum lipids in patients with type 2 diabetes // Clin. Lab. 2012. Vol. 58. P. 1283-1291.
9. Bajaj M., Suraamornkul S., Romanelli A., Cline G.W. et al. Effect of a sustained reduction in plasma free fatty acid concentration on intramuscular long-chain fatty acyl-coAs and insulin action in type 2 diabetic patients // Diabetes. 2005. Vol. 54, N 11. P. 3148-3153.
10. Cusi K., Kashyap S., Gastaldelli A. et al. Effect on insulin secretion and insulin action of a 48-h reduction of plasma free fatty acids with acipimox in nondiabetic subjects genetically predisposed to type 2 diabetes // Am. J. Physiol. Metab. 2007. Vol. 292. P. 1775-1778.
11. Brans C., Grunnet L.G. Mechanisms in endocrinology: Skeletal muscle lipotoxicity in insulin resistance and type 2 diabetes: a causal mechanism or an innocent bystander? // Eur. J. Endocrinol. 2017. Vol. 176, N 2. P. 67-78.
12. Samuel V.T., Shulman G. Mechanisms for insulin resistance: Common threads and missing links // Cell. 2012. Vol. 148. P. 852871.
13. Turinsky J., Bayly B.P., O’Sullivan D.M. 1,2-Diacylglycerol and ceramide levels in rat skeletal muscle and liver in vivo. Studies with insulin, exercise, muscle denervation, and vasopressin // J. Biol. Chem. 1990. Vol. 265, N 14. P 7933-7938.
14. Summers S.A. Ceramides in insulin resistance and lipotoxicity // Prog. Lipid Res. 2006. Vol. 45, N 1. P 42-72.
15. Veret J., Coant N., Berdyshev E.V., Skobeleva A. et al. Ceramide synthase 4 and de novo production of ceramides with specific N-acyl chain lengths are involved in glucolipotoxicity-induced apoptosis of INS-1 β-cells // Biochem. J. 2011 Vol. 438, N 1. P 177-189.
16. Boslem E., Weir J.M., MacIntosh G., Sue N. et al. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic β-cells// J. Biol. Chem. 2013. Vol. 288, N 37. P 26569-26582.
17. Shimabukuro M., Zhou Y.T., Levi M., Unger R.H. Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes // Proc. Natl. Acad. Sci USA. 1998. Vol. 95, N 5. P 2498-2502.
18. Engin A.B. What is lipotoxicity? Adv. Exp. Med. Biol. 2017; 960: 197-220. doi: 10.1007/978-3-319-48382-5_8
19. Walther T.C., Farese R.V. Jr. The life of lipid droplets // Biochim. Biophys. Acta. 2009. Vol. 1791, N 6. P 459-466.
20. Aon M.A., Bhatt N., Cortassa S.C. Mitochondrial and cellular mechanisms for managing lipid excess // Front. Physiol. 2014; 5: 282.
21. Lowell B.B., Shulman G.I. Mitochondrial dysfunction and type 2 diabetes // Science. 2005. Vol. 307. P 384-387.
22. Petersen K.F., Befroy D., Dufour S., Dziura J. et al. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance// Science. 2003. Vol. 300. P 1140-1142.
23. Gao D., Nong S., Huang X., Lu Y. et al. The effects of palmitate on hepatic insulin resistance are mediated by NADPH Oxidase 3-derived reactive oxygen species through JNK and p38MAPK pathways // J. Biol. Chem. 2010. Vol. 285, N 39. P. 29965-29973.
24. Aguirre V., Uchida T., Yenush L., Davis R., White M.F The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) // J. Biol. Chem. 2000. Vol. 275, N 12. P 9047-9054.
25. Ye R., Jung D.Y., Jun J.Y., Li J. et al. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance // Diabetes. 2010. Vol. 59 P 6-16.
26. Lee M.W., Chanda D., Yang J., Oh H. et al. Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH // Cell Metabolism 2010. Vol. 11, N 4. P. 331-339.
27. Seifert E.L., Estey C., Xuan J.Y., Harper M.E. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation // J. Biol. Chem. 2010. Vol. 285. P 5748-5758.
28. Ghosh J., Das J., Manna P, Sil PC. Taurine prevents arsenic-induced cardiac oxidative stress and apoptotic damage: Role of NF-kappa B, p38 and JNK MAPK pathway // Toxicol. Appl. Pharmacol. 2010. Vol. 240, N 1. P. 73-87.
29. Banhegyi G., Benedetti A., Csala M., Mandl J. Stress on redox // FEBS Letters. 2007. Vol. 581 P 3634-3640.
30. Lam T.K., Yoshii H., Haber C.A., Bogdanovic E. et al. Free fatty acid-induced hepatic insulin resistance: A potential role for protein kinase C-delta // Am. J. Physiol. Endocrinol. Metab. 2002. Vol. 283, N 4. P E682-E691.
31. Berg A.H., Combs T.P, Du X. et al. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action // Nat. Med. 2001. Vol. 7 P 947953.
32. Yamauchi T., Kamon J., Waki H. et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity // Nat. Med. 2001. Vol. 7. P 941-946.
33. Holland W.L., Miller R.A., Wang Z.V. et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin // Nat. Med. 2011. Vol. 17. P 55-63.
34. Xia J.Y., Holland W.L., Kusminski C.M. et al. Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis // Cell Metab. 2015. Vol. 22. P. 266278.
35. Qi Y., Takahashi N., Hileman S.M. et al. Adiponectin acts in the brain to decrease body weight // Nat. Med. 2004. Vol. 10. P 524529.
36. Kusminski C.M., McTernan P.G., Schraw T. et al. Adiponectin complexes in human cerebrospinal fluid: distinct complex distribution from serum // Diabetologia. 2007. Vol. 50. P 634-642.
37 Despres J.P., Golay A., Sjostrom L. Rimonabant in Obesity-Lipids Study Group: Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia // N. Engl. J. Med. 2005. Vol. 353. P 21212134.
38. Kadowaki T., Yamauchi T., Kubota N., Hara K. et al. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome // J. Clin. Invest. 2006. Vol. 116. P 1784-1792.
39. Snijder M.B., Heine R.J., Seidell J.C., Bouter L.M. et al. Associations of adiponectin levels with incident impaired glucose metabolism and type 2 diabetes in older men and women: the Hoorn study // Diabetes Care.
2006. Vol. 29. P. 2498-2503.
40. Аметов А.С. Ожирение. Современный взгляд на патогенез и терапию. Т. 1. М. : ГЭОТАР-Медиа, 2019. 384 с.
41. Воробьев С.В., Петровская Е.Ю., Кузьменко Н.А., Хрипун И.А. Новый препарат в комплексной терапии сахарного диабета. Пострегистрационный опыт применения у пациентов с сахарным диабетом 1-го и 2-го типа // Медицинский совет. 2018. № 16. С. 28-34.
42. Gorbunov E.A., Nicoll J., Myslivets A.A., Kachaeva E.V., Tarasov S.A. Subetta enhances sensitivity of human muscle cells to insulin // Bull. Exp. Biol. Med. 2015. Vol. 159, N 4. P 463-465.
43. Gorbunov E.A., Nicoll J., Kachaeva E.V., Tarasov S.A., Epstein O.I. Subetta increases phosphorylation of insulin receptor β-subunit alone and in the presence of insulin. Nutr. Diabetes. 2015. Vol. 5, N 7. P e169. doi:10.1038/nutd.2015.20
44. Белоус А.С., Покровская Т.Г., Покровский М.В. и др. Изучение кардиопротективных эффектов смеси гомеопатических разведений поликлональных кроличьих антител к эндотелиальной синтазе оксида азота (eNOS) С12, С30, С200 при экспериментальном моделировании L-NAME индуцированного дефицита оксида // Тезисы докладов XIV Российского национального конгресса “Человек и лекарство”. М., 2007.
45. Покровский М.В., Кочкаров В.И., Покровская Т.Г. и др. Сравнительное изучение потенциальных эндотелийпротекторов и препарата Импаза при моделировании дефицита оксида азота // Бюл. экспер. биол. 2009. Т. 148, № 8, прил. С. 154-158.
46. Nicoll J., Gorbunov E.A., Tarasov S.A., Epstein O.I. Subetta treatment increases adiponectin secretion by mature human adipocytes in vitro // Int. J. Endocrinol. 2013. Vol. 2013. P 925874.
References
1. Dedov I.I., Shestakova M.V., Vikulova O.K. Epidemiology of diabetes the federal diabetes registry. Sakharni diabet [Diabetes Mellitus]. 2017; mellitus in Russian Federation: clinical and statistical report according to 20 (1): 13-41. (in Russian)
2. The Ministry of Health called the regions suffering from obesity https://www.rbc.ru/society/24/07/2018/5b519ee49a7947f2d4d7fa9b (in Russian)
3. Hajer G.R., wan Haeften T.W., Visseren F.L. Adipose tissue disfunction in obesity, diabetes and vascular diseases. Eur Heart J. 2008; 29 (24): 2959-71. doi: 10.1093/eurheartj/ehn387
4. Dedov 1.1., Melnichenko G.A., Butrova S.A. Adipose tissue as an endocrine organ. Ozhirenie i metabolism [Obesity and Metabolism]. 2006; (1): 6-13. (in Russian)
5. Guo Z., Hensrud D.D., Johnson C.M., Jensen M.D. Regional postprandial fatty acid metabolism in different obesity phenotypes. Diabetes.1999; 48 (8): 1586-92.
6. Ebbert J.O., Jensen M.D. Fat depots, free fatty acids, and dyslipidemia. Nutrients. 2013; 5 (2): 498-508.
7. Unger R.H. Lipotoxic diseases. Ann Rev Med. 2002; 53: 319-36.
8. Korani M., Firoozrai M., Maleki J., et al. Fatty acid composition of serum lipids in patients with type 2 diabetes. Clin Lab. 2012; 58 (1112): 1283-91.
9. Bajaj M., Suraamornkul S., Romanelli A., Cline G.W., et al. Effect of a sustained reduction in plasma free fatty acid concentration on intramuscular long-chain fatty acyl-coAs and insulin action in type 2 diabetic patients. Diabetes. 2005; 54 (11): 3148-53.
10. Cusi K., Kashyap S., Gastaldelli A., et al. Effect on insulin secretion and insulin action of a 48-h reduction of plasma free fatty acids with acipimox in nondiabetic subjects genetically predisposed to type 2 diabetes. Am J Physiol Metab. 2007; 292: 1775-8.
11. Brans C., Grunnet L.G. Mechanisms in endocrinology: Skeletal muscle lipotoxicity in insulin resistance and type 2 diabetes: a causal mechanism or an innocent bystander? Eur J Endocrinol. 2017; 176 (2): 67-78.
12. Samuel V.T., Shulman G. Mechanisms for insulin resistance: Common threads and missing links. Cell. 2012; 148: 852-71.
13. Turinsky J., Bayly B.P., O’Sullivan D.M. 1,2-Diacylglycerol and ceramide levels in rat skeletal muscle and liver in vivo. Studies with insulin, exercise, muscle denervation, and vasopressin. J Biol Chem. 1990; 265 (14): 7933-8.
14. Summers S.A. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006; 45 (1): 42-72.
15. Veret J., Coant N., Berdyshev E.V., Skobeleva A., et al. Ceramide synthase 4 and de novo production of ceramides with specific N-acyl chain lengths are involved in glucolipotoxicity-induced apoptosis of INS-1 β-cells. Biochem J. 2011; 438 (1): 177-89.
16. Boslem E., Weir J.M., Macintosh G., Sue N., et al. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic β-cells. J Biol Chem. 2013; 288 (37): 26569-82.
17. Shimabukuro M., Zhou Y.T., Levi M., Unger R.H. Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci USA. 1998; 95 (5): 2498-502.
18. Engin A.B. What is lipotoxicity? Adv Exp Med Biol. 2017; 960: 197-220. doi: 10.1007/978-3-319-48382-5_8
19. Walther T.C., Farese R.V. Jr. The life of lipid droplets. Biochim Biophys Acta. 2009; 1791: 459-66.
20. Aon M.A., Bhatt N., Cortassa S.C. Mitochondrial and cellular mechanisms for managing lipid excess. Front Physiol. 2014; 5: 282.
21. Lowell, B.B., and G.I. Shulman. Mitochondrial dysfunction and type 2 diabetes. Science. 2005; Vol. 307: 384-7.
22. Petersen, K.F., Befroy D., Dufour S., Dziura J., et al. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science. 2003; 300: 1140-2.
23. Gao D., Nong S., Huang X., Lu Y., et al. The effects of palmitate on hepatic insulin resistance are mediated by NADPH Oxidase 3-derived reactive oxygen species through JNK and p38MAPK pathways. J Biol Chem. 2010; 285 (39): 29965-73.
24. Aguirre V., Uchida T., Yenush L., Davis R., White M.F. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem. 2000; 275 (12): 9047-54.
25. Ye R., Jung D.Y., Jun J.Y., Li J., et al. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes. 2010; 59: 6-16.
26. Lee M.W., Chanda D., Yang J., Oh H., et al. Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metabolism. 2010; 11 (4): 331-9.
27. Seifert E.L., Estey C., Xuan J.Y., Harper M.E. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J Biol Chem. 2010; 285: 5748-58.
28. Ghosh J., Das J., Manna P., Sil P.C. Taurine prevents arsenic-induced cardiac oxidative stress and apoptotic damage: Role of NF-kappa B, p38 and JNK MAPK pathway. Toxicol Appl Pharmacol. 2010; 240: 73-87.
29. Banhegyi G., Benedetti A., Csala M., Mandl J. Stress on redox. FEBS Letters. 2007; 581: 3634-40.
30. Lam T.K., Yoshii H., Haber C.A., Bogdanovic E., et al. Free fatty acid-induced hepatic insulin resistance: A potential role for protein kinase C-delta. Am J Physiol Endocrinol Metab. 2002; 283 (1): E682-91.
31. Berg A.H., Combs T.P., Du X., et al. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med. 2001; 7: 947-53.
32. Yamauchi T., Kamon J., Waki H., et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001; 7: 941-6.
33. Holland W.L., Miller R.A., Wang Z.V., et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011; 17: 55-63.
34. Xia J.Y., Holland W.L., Kusminski C.M., et al. Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis. Cell Metab, 2015; 22: 266-78.
35. Qi Y., Takahashi N., Hileman S.M. et al. Adiponectin acts in the brain to decrease body weight. Nat Med. 2004; 10: 524-9.
36. Kusminski C.M., McTernan P.G., Schraw T., et al. Adiponectin complexes in human cerebrospinal fluid: distinct complex distribution from serum. Diabetologia. 2007; 50: 634-42.
37. Despres J.P., Golay A., Sjostrom L. Rimonabant in Obesity-Lipids Study Group: Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med. 2005; 353: 2121-34.
38. Kadowaki T., Yamauchi T., Kubota N., Hara K., et al. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006; 116: 1784-92.
39. Snijder M.B., Heine R.J., Seidell J.C., Bouter L.M., et al. Associations of adiponectin levels with incident impaired glucose metabolism and type 2 diabetes in older men and women: the Hoorn study. Diabetes Care. 2006; 29: 2498-503.
40. Ametov A.S. Obesity. Modern view on pathogenesis and therapy. Vol. 1. Moscow: GEOTAR-Media, 2019: 384 p. (in Russian)
41. Vorob’yev S.V., Petrovskaya E.U., Kuz’menko N.A., Khripun I.A. А new drug in the complex therapy of diabetes mellitus. post-registration experience in patients with type 1 and type 2 diabetes mellitus. Meditsinskiy sovet [Medical Council]. 2018; (16): 28-34. (in Russian)
42. Gorbunov E.A., Nicoll J., Myslivets A.A., Kachaeva E.V., Tarasov S.A. Subetta enhances sensitivity of human muscle cells to insulin. Bull Exp Biol Med. 2015; 159 (4): 463-5.
43. Gorbunov E.A., Nicoll J., Kachaeva E.V., Tarasov S.A., Epstein O.I. Subetta increases phosphorylation of insulin receptor β-subunit alone and in the presence of insulin. Nutr. Diabetes. 2015. Vol. 5, N 7. P e169. doi:10.1038/nutd.2015.20
44. Belous A.S., Pokrovskaya T.G., Pokrovsky M.V., et al. Studying of cardioprotective effects of mix of homeopathic dilutions of polyclonal rabbit antibodies to endothelial synthase of nitrogen oxide (eNOS) C12, C30, C200 at experimental modeling of L-NAME of the induced deficiency of oxide. In: Abstracts of the reports of reports of the XIV Russian National Congress “Man and Drug”. Moscow; 2007. (in Russian)
45. Pokrovsky M.V., Kochkarov V.I., Pokrovskaya T.G., et al. Comparative study of potential endothelium protectors and Impaza drug in nitric oxide deficiency simulation. Byulleten’ eksperimental’noy biologii i meditsiny [Bulletin of Experimental Biology and Medicine]. 2009; 148 (8; Suppl): 154-8. (in Russian)
46. Nicoll J., Gorbunov E.A., Tarasov S.A., Epstein O.I. Subetta treatment increases adiponectin secretion by mature human adipocytes in vitro. Int J Endocrinol. 2013; 2013: 925874.