В мире в настоящее время более 400 миллионов человек страдают сахарным диабетом, и более того, прогнозируется рост заболеваемости (в основном за счет сахарного диабета 2 типа). Для этой патологии характерно нарушение гомеостаза глюкозы в крови. Сахарный диабет типа 1, на который приходится около 7–12 % зарегистрированных случаев, является хроническим аутоиммунным заболеванием, вызванным разрушением инсулин-продуцирующих панкреатических β-клеток. Дефицит секреции инсулина значительно снижает способность мышечной и жировой ткани накапливать глюкозу в виде гликогена. Это в сочетании с усилением глюконеогенеза способствует развитию хронической гипергликемии.
В патогенезе сахарного диабета 2 типа, на долю которого приходится 87–91 % случаев, ключевую роль играют резистентность к инсулину и дисфункция β-клеток поджелудочной железы. Инсулинорезистентность способствует активации выработки эндогенной глюкозы в печени. Компенсаторное увеличение секреции β-клетками инсулина приводит в итоге к их «перегрузке», формируется относительная недостаточность инсулина. Гипергликемия также усугубляет повреждение β-клеток ввиду токсичности глюкозы.
У 20–40 % пациентов с сахарным диабетом диагностируют хроническую болезнь почек (ХБП). Морфологически чаще всего отмечается утолщение базальной мембраны в клубочках, расширение мезангиального матрикса, нодулярный гломерулосклероз и гиалиноз артериол. В тубулоинтерстициальном пространстве наблюдается накопление структур внеклеточного матрикса, утолщение базальной мембраны, воспаление (полиморфноклеточная инфильтрация). Функционально это приводит к изменению скорости клубочковой фильтрации (СКФ): начальная гиперфильтрация с последующим постепенным снижением СКФ и увеличение экскреции белка с мочой (на начальных стадиях — микроальбуминурия).
Наличие ХБП также значительно увеличивает вероятность сердечно-сосудистых заболеваний и смерти [1].
Почки являются важным регулятором гомеостаза глюкозы в крови. Их вклад был признан еще в 1938 году. Они не только активно реабсорбируют отфильтрованную глюкозу, направляя ее обратно в кровеносное русло, но также синтезируют глюкозу посредством глюконеогенеза. Кроме того, в различных отделах почек происходит синтез глюкозы de novo из жирных кислот. Почки активно потребляют «глюкозное топливо» как через окислительное фосфорилирование, так и через гликолитические пути (рис. 1) [2].
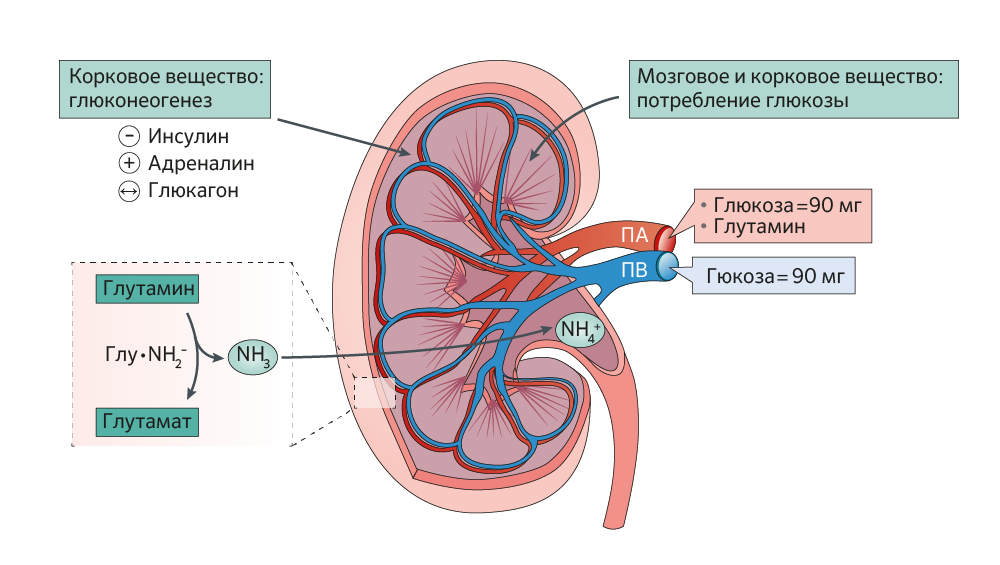
В почечной паренхиме осуществляется весь спектр превращений молекул глюкозы: не только процессы реабсорбции, но и глюконеогенез и использование глюкозы как энергетического материала.
Гормональная регуляция эндогенной продукции глюкозы: инсулин тормозит синтез глюкозы путем снижения доступности субстратов, подавления синтеза ферментов, одновременно стимулируя гликогеногенез и гликолиз; глюкагон стимулирует глюконеогенез и гликогенолиз в печени, влияние на почечный глюконеогенез не подтверждено; катехоламины увеличивают глюконеогенез в почках, снижают поглощение глюкозы тканями, ингибируют секрецию инсулина, стимулируют секрецию глюкагона. ПА-почечная артерия, ПВ – почечная вена [2].
Метаболизм глюкозы
Глюкоза из кровеносного русла в виде небольшой молекулы свободно фильтруется клубочками, которые являются ключевым компонентом системы фильтрации почек. При СКФ 180л/сут. и средней концентрации глюкозы в плазме 5,5 ммоль/л ежедневная почечная реабсорбция (обратное всасывание) глюкозы составляет 180 г. Далее глюкоза поступает в просвет канальцев, где судьбой всех отфильтрованных молекул является либо реабсорбция обратно в кровоток, либо экскреция с мочой. Если при развитии гипергликемии почечный порог глюкозы превышен, возникает глюкозурия.
Когда концентрация глюкозы в крови находится в пределах физиологического диапазона 4,0–7,8 ммоль/л, фактически вся отфильтрованная глюкоза реабсорбируется, поскольку потеря глюкозы с мочой энергетически неэффективна. Большая часть отфильтрованной глюкозы (от 83 до 98 %) реабсорбируется из просвета проксимальных извитых канальцев посредством активного транспорта через натрий-глюкозный котранспортер 2 типа (SGLT2), расположенный на апикальной мембране эпителиальных клеток. Этот тип транспортеров обладает низкой аффинностью, но значительной емкостью (1 молекула натрия на 1 молекулу глюкозы) и представлен практически только в эпителиоцитах начального отдела проксимального канальца нефрона. Небольшое количество глюкозы, которое попадает в проксимальные прямые канальцы, поглощается с помощью натрий-глюкозного котранспортера 1 типа (SGLT1), который имеет высокую аффинность при низкой емкости (2 молекулы натрия на 1 молекулу глюкозы). Транспортер 1 типа также осуществляет процессы реабсорбции глюкозы и галактозы в кишечнике.
Поглощенная глюкоза высвобождается обратно в кровоток посредством пассивного транспорта через клеточную мембрану с помощью глюкозных транспортеров GLUT1 и GLUT2 на базолатеральной мембране. Также в клубочках, проксимальном канальце и толстой части восходящего колена петли Генле был обнаружен подтип GLUT4.
Перенос молекулы глюкозы внутрь эпителиоцита проходит с затратой энергии против градиента концентрации. Обратный транспорт ионов Na+ из клетки осуществляется Na+K+-АТФазой. Ее работа сопряжена с использованием огромного количества энергии (примерно 50 % от суммарной потребности почек в АТФ в физиологических условиях). При высокой гликемии увеличивается почечная реабсорбция глюкозы, а с ней и абсорбция Na+, для возврата которого в кровоток не хватает энергии. Концентрация ионов Na+ снижается, клетки юкстагломерулярного аппарата улавливают это, и происходит выброс ренина, который повышает артериальное давление и далее способствует замедлению клубочковой фильтрации.
Повышенная реабсорбция глюкозы в проксимальных канальцах у пациентов с сахарным диабетом достигается за счет увеличения экспрессии транспортера SGLT2 на поверхности эпителиоцитов. Это поддерживает высокий уровень гликемии, воздействовать на который призваны гипогликемические препараты из класса селективных ингибиторов SGLT2 — глифлозины, понижающие реабсорбцию за счет обратимой блокады транспортной системы на фоне нарастания экскреции глюкозы [3].
Синтез глюкозы
В печени и корковом веществе почек в постабсорбтивный период (через 14–16 часов после приема пищи) посредством глюконеогенеза синтезируется эквивалентное количество глюкозы. Основным субстратом в обоих органах является лактат, но в его отсутствии почки «выбирают» глютаминовую кислоту, печень — аланин. Вклад печени в постпрандиальный уровень гликемии (через 4–6 ч после приема пищи) в физиологических условиях увеличивается за счет гликогенолиза, и составляет в итоге 80 %. По мере деградации большей части гликогена в печени нарастает доля почечного глюконеогенеза, которая может составлять до 60 % эндогенной продукции глюкозы в организме в постпрандиальный период.
Повышение уровня циркулирующего инсулина снижает выработку эндогенной глюкозы, при этом гликогенолиз прекращается, а печеночный глюконеогенез снижается на 82 %. Как и в случае с гепатоцитами, инсулин также подавляет синтез и высвобождение глюкозы в почках в норме. Однако при сахарном диабете, когда эффект инсулина снижен или отсутствует, наблюдается значительное увеличение почечного глюконеогенеза. Данный процесс возможен за счет накопления гликогена, которое отмечается в «диабетических почках».
Кроме того, почки также непосредственно участвуют в инактивация 30–40 % молекул инсулина (6–8 ЕД/сут). Деградация инсулина происходит внутри почечных эпителиоцитов при участии инсулиновой протеазы, глютатион-инсулин трансгидрогеназы, а также лизосомальных ферментов. При ХБП и нарастании почечной недостаточности увеличивается период полураспада инсулина, что снижает потребность таких пациентов в экзогенном инсулине [4].
Использование глюкозы
В клетках почечных канальцев отмечается большое количество митохондрий, что объясняет их высокое энергопотребление. Значительное число молекул АТФ, генерируемого посредством окислительного фосфорилирования, делает почки основным потребителем молекулярного кислорода, уступая только миокарду в состоянии покоя. Предпочтительное топливо для клеток почек различается в зависимости от их расположения вдоль нефрона, их специфических потребностей в АТФ и доступности кислорода. Например, в то время как клубочки и тонкая дистальная часть являются «сладкоежками» и предпочитают глюкозу, клетки проксимальных канальцев, как правило, используют свободные жирные кислоты (СЖК). Для окисления СЖК требуется больше кислорода, но выход АТФ на грамм выше, что делает его приоритетным в условиях гипероксии.
На начальных этапах развития диабета потребление кислорода почками резко увеличивается. В условиях повышенной потребности и невозможности ее обеспечения, формируется гипоксия ткани, и в конечном итоге переключение на гликолиз с увеличением реакций окисления глюкозы даже в проксимальных канальцах, в норме использующих СЖК. А клетки, и ранее получавшие энергию путем гликолиза, теперь нуждаются в еще большем количестве глюкозы, при этом главные «сладкоежки» располагаются в толстой части восходящего колена петли Генле [5].
Аномальное накопление гликогена в почках, отмечаемое при сахарном диабете, может быть результатом этого перехода на «глюкозную топливную систему». Возможно, это адаптивный механизм, направленный на сохранения почечной функции. Но далее, с прогрессированием ХБП, гликоген способствует нарастанию структурного повреждения почек [6].
Список литературы:
- Anders H.J., Huber T.B., Isermann B., Schiffer M. CKD in diabetes: diabetic kidney disease versus nondiabetic kidney disease. Nature Reviews Nephrology. 2018;14(6):361.
- DeFronzo R.A., et al. Renal, metabolic and cardiovascular considerations of SGLT2 inhibition. Nat Rev Nephrol. 2017;13(1):11-26.
- Alsahli M., Gerich J.E. Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes research and clinical practice. 2017;133:1-9.
- Sullivan, Mitchell A. et al.Glucose and glycogen in the diabetic kidney: Heroes or villains? EBioMedicine. Published online: August 10, 2019
- Forbes JM, Thorburn DR. Mitochondrial dysfunction in diabetic kidney disease. Nature Reviews Nephrology. 2018 May;14(5):291.
- Jiang, W., Xiao, T., Han, W., Xiong, J., He, T., Liu, Y. et al. Klotho inhibits PKCα/p66SHC-mediated podocyte injury in diabetic nephropathy. Mol Cell Endocrinol. 2019; 494: 11049
источник: medach.pro