Способность адипоцитов вырабатывать тепловую энергию может оказаться ключом в борьбе с метаболическими нарушениями. Несмотря на широкую известность агонистов β3-адренорецепторов как индукторов термогенеза, побочным эффектом их активации является риск развития сердечно-сосудистых заболеваний. Согласно результатам нового исследования, рецептор третьего типа, сопряженный с G-белком, способен осуществлять управление термогенезом жировой ткани независимо от сигнализации посредством β3-адренорецепторов.
Классический способ индуцирования программирования термогенеза в адипоцитах, опосредованный рецепторами, сопряженными с G-белком (GPCR, от англ. G-protein coupled receptors), включает β-адренорецепторы трех типов (ADRB1, ADRB2 и ADRB3). При связывании лиганда (например, норадреналина) β-адренергические рецепторы опосредуют Gs-белок-сопряженное производство цАМФ при помощи аденилилциклазы 1 (см. рис. 1а). Это повышение уровня цАМФ приводит к активации путей, участвующих в захвате глюкозы, β-окислении, мобилизации липидов и разобщении цепи переноса электронов, дабы в конечном итоге преобразовать химическую энергию в тепло [2]. Теоретически, модуляция этого пути при помощи агонистов β3-адренергических рецепторов может показаться возможной стратегией противодействия метаболическим нарушениям. Поскольку экспрессия гена Adrb3 наблюдается преимущественно в адипоцитах, долгое время считалось, что его стимуляция приведет к уменьшению массы жировой ткани с ограниченным количеством побочных эффектов [1]. К сожалению, фармакологические активаторы β3-адренорецепторов, такие как мирабегрон, способствуют повышению артериального давления, и их прием в клинически эффективных дозах связан с развитием сердечно-сосудистых рисков, что исключает их широкое использование [2]. При метаболических нарушениях этот сердечно-сосудистый риск лишь возрастает [3]. Таким образом, исследование, проведенное в 2020 году на людях, свидетельствует о важной роли альтернативных механизмов, например, β2-адренергической стимуляции, для термогенеза и липолиза жировой ткани [4]. Однако слишком рано делать выводы о некоем конкретном клиническом применении результатов этих исследований, так как еще крайне мало известно об альтернативных способах индукции термогенеза по сравнению с агонистами β3-адренергических рецепторов.
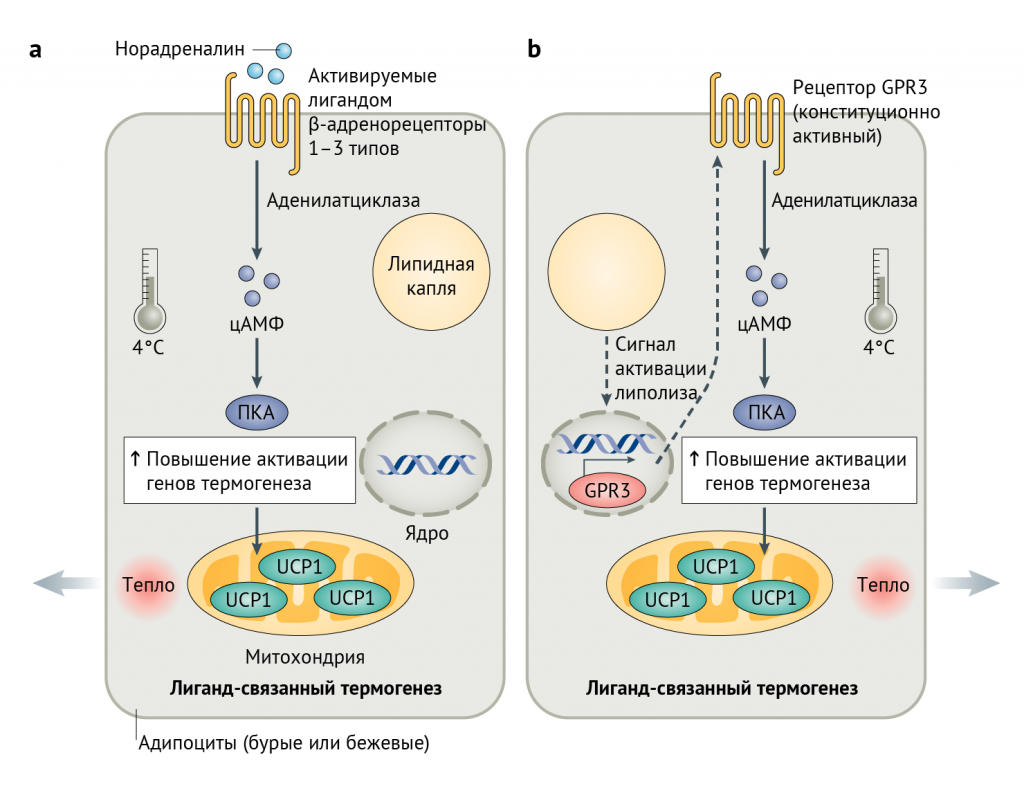
Упрощенная схема, изображающая ключевые различия между классическим путем активации термогенеза (часть a) и внутренне-обусловленным программированием термогенеза, опосредованным G-белок-связанным-рецептором 3 (GPR3) (часть b).
а | Низкая температура вызывает высвобождение норадреналина (посредством симпатической нервной системы), который связывается с β-адренорецепторами, что стимулирует выработку цАМФ, что приводит к усилению экспрессии генов термогенеза посредством протеинкиназы А (ПКА).
b | Механизм, открытый Свейдалом Йохансеном, демонстрирует, что лиганд для GPCR не нужен. Скорее всего, не установленный до сих пор липолитический сигнал в значительной степени увеличивает экспрессию GPR3, который способствует продукции цАМФ за счет присущей ему N-концевой активности.
ADRB1–3 — β-адренорецепторы;
UCP1 — разобщающий белок 1 (см. объяснение в тексте).
Предыдущая работа была сосредоточена на открытии неканонических путей активации термогенных адипоцитов [3]; однако Свейдал Йохансен с соавт. заявляют, что необходимо признать важность факторов, влияющих на транскрипционный контроль самих GPCR [2]. Внутренне обусловленная и не связанная с лигандом роль GPCR часто не принимается во внимание, как и факторы, которые модулируют экспрессию этих ключевых компонентов плазматической мембраны. GPR3, GPR6 и GPR12 давно признаны конституционно активными GPCR, экспрессия которых в центральной нервной системе достигает высоких значений [5]. GPR3, например, является ключевым для неврологических процессов (рост аксонов и синтез амилоида-β5). Однако ученые лишь подходят к описанию роли тех факторов, которые влияют на контроль транскрипции этих конституционно активных GPCR6.
В своем исследовании Свейдал Йохансен с соавт. продемонстрировали (на мышиных моделях метаболических заболеваний и у людей), что экспрессия гена Gpr3 индуцируется низкой температурой посредством молекулярного пути сигнализации липолиза (не являющегося классическим), который достаточно эффективно запускает термогенез как в бурых, так и в бежевых адипоцитах [2]. Используя метод прицельной количественной ПЦР, авторы исследовали 44 Gs-белок-сопряженных рецептора у мышей в комфортных условиях (29 °C) и после воздействия холода (4 °C). В результате обнаружилось, что GPR3 оказались той группой холодовых рецепторов, которая в наибольшей степени индуцировалась вследствие воздействия низких температур (в подкожной белой жировой ткани и бурой жировой ткани [БЖТ]) [2]. Действительно, в этих термогенных жировых депо наблюдалась повышенная регуляция экспрессии как самого гена, так и белка. Чтобы подтвердить отсутствие связи между сигнализацией посредством GPR3 и β-адренергической сигнализацией, авторы приглушили этот путь сигнализации. Последнее было реализовано посредством использования в эксперименте мышей, у которых была произведена генетическая абляция генов Adrb1, Adrb2 и Adrb3 (в итоге эти животные были лишены β-адренорецепторов). Исследователи отметили дополнительное увеличение экспрессии GPR3 как в условиях комнатной температуры, так и при низкой температуре, что лишний раз подтверждает важность данного типа GPCR в жировой ткани мышей [2].
Чтобы продемонстрировать роль GPR3 в условиях in vivo, авторы создали животную модель, характеризовавшуюся сверхэкспрессией Gpr3 в бурых и бежевых адипоцитах, и обнаружили, что у этих мышей была значительно более низкая масса тела, чем у животных группы контроля (различие было статистически значимым) [2]. Это открытие оказалось верным не только для подкожной белой жировой ткани и БЖТ, но также и для белой жировой ткани придатка яичка, в которой нет признаков опосредованного белком термогенином (т. н. UCP1 — от англ. uncoupling protein 1 — разобщающий белок первого типа) термогенеза [2]. В условиях питания продуктами с высоким содержанием жиров у трансгенных мышей со сверхэкспрессией GPR3 в термогенных жировых депо риск развития алиментарного ожирения оказался значительно снижен, невзирая на сохранение прежнего суточного калоража [2]. Более того, при переходе на диету с высоким содержанием жиров расход энергии у трансгенных мышей увеличился, а коэффициент дыхательного газообмена снизился. Согласно этому, предполагается, что липиды в продуктах питания используются в качестве топлива для термогенеза, опосредованного GPR3 [2]. По сравнению с животными группы контроля, у этих мышей обнаружилась повышенная толерантность к глюкозе, а в печени отсутствовало эктопическое отложение липидов [2]. Интересно, что эти результаты основаны на выводах предыдущего исследования, в котором было продемонстрировано, что у мышей, характеризовавшихся генетическим нокаутом Gpr3 во всем организме, была обнаружена сниженная экспрессия генов термогенеза, а у самих животных была выявлена склонность к ожирению в преклонном для них возрасте [7]. Чтобы изучить связь с ожирением у людей, Свейдал Йохансен с соавт. определили экспрессию GPR3 в надключичной БЖТ у добровольцев. В результате было отмечено, что более высокая экспрессия этого рецептора оказалась связана с более низким ИМТ у лиц, толерантных к глюкозе, по сравнению с образцами, взятыми у лиц с нарушенным контролем концентрации глюкозы в крови [2]. Это открытие было подкреплено исследованиями бурых адипоцитов человека in vitro, в которых выключение гена, кодирующего GPR3, резко снижало экспрессию UCP1 [2].
Напротив, когда исследователи применили методику CRISPR-Cas9 для управления экспрессией GPR3 в подкожных белых адипоцитах человека, обнаружилось усиление захвата жирных кислот и ускорение работы дыхательной цепи митохондрий, а также усиление индукции термогенных генов, таких как гены белков UCP1 и PRDM16. Согласно этим результатам, экспрессия GPR3 может стимулировать потемнение адипоцитов сама по себе, посредством альтернативного хорошо известного пути, опосредованного норадреналином. Хотя GPR3-опосредованное термогенное программирование, описанное в настоящем исследовании, не является лиганд-зависимым, в действительности оно осуществляется посредством пока не идентифицированного сигнала о запуске липолиза (см. рис. 1b), поскольку двойное отключение генов липазы триглицеридов жиров и гормон-чувствительной липазы значительно снижает индуцированную холодом экспрессию GPR3 [2]. Это открытие согласуется с другими работами, демонстрирующими, что отключение липолиза на генетическом уровне снижает активацию термогенеза [8, 9]. Хотя в этой работе продемонстрирована внутренне обусловленная способность GPR3 запускать термогенез, целями будущих исследований должны стать выяснение природы липолитического сигнала (или сигналов), влияющих на контроль транскрипции. Например, в исследовании 2018 года было показано, что 130 видов липидов претерпели изменения в БЖТ у мышей, испытавших воздействие низкой температуры [10]. В частности, было выявлено, что активизировался метаболизм фосфатидилглицерина и кардиолипина; данное открытие является почвой для дальнейшего изучения [10]. Существует также вопрос о том, к запуску какого сигнального пути приводит липолитический сигнал для индукции экспрессии GPR3, поскольку Свейдал Йохансен с соавт. применили выключение генов PPARα, PPARγ и известных липид-активируемых ядерных рецепторов в коричневых адипоцитах посредством малых интерферирующих РНК, однако норадреналин-индуцированная активация экспресии гена Gpr3 не была устранена (ссылка 2). Захватывающие ответы на эти вопросы могут послужить еще большим подтверждением идеи о том, что точечное воздействие на GPR3 посредством активаторов липолиза является вполне реализуемой стратегией смягчения выраженности протекания нарушений метаболизма.