Аннотация
Ожирение – сложная и актуальная медико-социальная проблема мирового уровня. Жировая ткань является не только местом депонирования энергетических субстратов, но и источником секреции провоспалительных и противовоспалительных медиаторов, участвующих в развитии хронического латентного системного воспалительного процесса в организме при ожирении. Метаболический сигнал при ожирении способствует поляризации макрофагов в М1-направлении и запуску иммунного ответа по Тh1-типу, вызывая развитие хронического воспаления в жировой ткани. Хроническое воспалительное состояние играет ключевую роль в патофизиологии инсулинорезистентности, индуцированной ожирением. Возможным патофизиологическим звеном развития инсулинорезистентности при воспалении могут быть толл-подобные рецепторы (TLRs). В то же время индуцированный воспалением липолиз необходим для высвобождения энергетических ресурсов во время развития инфекционного процесса. Таким образом, низкоуровневое вялотекущее хроническое воспаление важно для защиты от дисфункции адипоцитов. Эти результаты позволяют предположить, что провоспалительная сигнализация не является исключительно патогенной при ожирении. В связи с этим особенно актуальным является изучение воспалительных сигнальных путей, участвующих в модуляции хронического воспаления жировой ткани. В данном обзоре суммированы современные взгляды на структуру, функцию TLRs и их участие в патогенезе хронического воспаления при ожирении. Обсуждается возможность использования TLRs в качестве терапевтической мишени при данной патологии. Очевидно, что дальнейшее изучение воспалительных сигнальных путей с участием TLRs, инициирующих развитие хронического воспаления жировой ткани, позволит разработать новые и эффективные терапевтические стратегии в отношении ожирения и его метаболических осложнений.
Ожирение – серьезная медико-социальная проблема мирового уровня, приобретающая глобальный масштаб в связи с увеличением показателей распространенности данной патологии как в странах с высоким уровнем жизни, так и в развивающихся странах [1]. По данным Всемирной организации здравоохранения (ВОЗ), в мире насчитывается 300–475 млн человек, страдающих ожирением. Прогнозируется, что к 2025 г. ожирение будет диагностировано у одной пятой части лиц трудоспособного возраста [2]. Увеличение числа больных с ожирением сопровождается возрастанием размера ущерба для мировой экономики. Ежегодные расходы на лечение данной патологии составляют 2 трлн долларов США. В Российской Федерации аналогичные затраты составляют около 369 млрд руб. (70% общих затрат бюджета) [3].
Особую проблему представляют метаболические осложнения ожирения и сочетанные с ним заболевания, существенно повышающие мировые показатели заболеваемости и сердечно-сосудистой смертности [4, 5]. В 2015 г. зарегистрировано 417 115 смертей и 14 448 548 лет потерь DALYs (Disability Adjusted Life Year – «год жизни, измененный или потерянный в связи с нетрудоспособностью»), связанных с ожирением, что составляет около 10% от общего числа смертей и 6,3% от потерь DALYs для лиц всех возрастных групп [6].
Абдоминальный тип ожирения является ключевым звеном в процессе формирования метаболического синдрома (МС) [7]. Распространенность МС среди взрослого населения развитых стран составляет 20–25% и имеет устойчивую тенденцию роста данного показателя [8]. Увеличение массовой доли висцерального жира сочетается со снижением чувствительности периферических тканей к инсулину и развитием гиперинсулинемии, что сопровождается нарушениями углеводного, липидного и пуринового обменов и развитием артериальной гипертензии [9, 10]. Все составляющие МС имеют общий этиопатогенетический механизм – воспаление. Жировая ткань является не только местом депонирования энергетических субстратов, но и источником секреции провоспалительных и противовоспалительных медиаторов, участвующих в развитии хронического латентного системного воспалительного процесса в организме при ожирении [11]. Хроническое воспалительное состояние играет ключевую роль в патофизиологии сахарного диабета 2 типа и инсулинорезистентности (ИР), индуцированной ожирением [12]. Метаболический сигнал при ожирении способствует поляризации макрофагов в М1-направлении (провоспалительный тип макрофагов) и запуску иммунного ответа по Тh1 типу, вызывая развитие хронического воспаления в жировой ткани [13]. Возможным патофизиологическим звеном развития ИР при воспалении могут быть толл-подобные рецепторы (TLRs) [14]. Именно данные рецепторы являются связующим звеном между врожденным, адаптивным иммунитетом и клеточным повреждением [15]. Усиление липолиза в адипоцитах приводит к повышению уровня ненасыщенных жирных кислот, которые посредством TLRs способствуют дифференцировке макрофагов в провоспалительный М1-фенотип [16]. Очевидно, что дальнейшее изучение воспалительных сигнальных путей с участием TLRs, инициирующих развитие хронического воспаления жировой ткани, позволит разработать новые и эффективные терапевтические стратегии в отношении ожирения и его метаболических осложнений.
Таким образом, ожирение представляет собой масштабную медико-социальную проблему мирового уровня, требующую пристального внимания. Несмотря на прогресс в изучении этиопатогенеза ожирения и достижения в разработке методов предотвращения развития и лечения данного заболевания, нерешенность существующей проблемы привела к заявлению ВОЗ о необходимости прекращения пандемии к 2025 г. [17].
В данном обзоре суммированы современные взгляды на структуру, функцию TLRs и их участие в патогенезе хронического воспаления при ожирении. Обсуждается возможность использования TLRs в качестве терапевтической мишени при данной патологии.
В базе данных PubMed осуществлялся поиск научных публикаций по данной теме за последние 10 лет. В обзор включали источники информации, в которых освещались вопросы структуры, функционирования и роли TLRs в патогенезе ожирения. Информационные запросы включали следующую совокупность ключевых слов: «toll-like receptors; obesity; insulin resistance; inflammation; adipose tissue». Найденные по запросу статьи просматривали на предмет их соответствия выбранным критериям включения и при положительном результате проводили анализ текста.
ХРОНИЧЕСКОЕ ВОСПАЛЕНИЕ ПРИ ОЖИРЕНИИ
Многие клинические и экспериментальные исследования позволили установить связь между висцеральным ожирением и хронической воспалительной реакцией в жировой ткани больных ожирением, ответственной за его метаболические осложнения [18–21].
Воспаление – типовой общепатологический процесс, одним из инициаторов которого являются сахара, белки, бактериальные липополисахариды (LPS) и липоолигосахариды, известные как патоген-ассоциированные молекулярные структуры (pathogen-associated molecular pattern (PAMPs)) [22]. Кроме того, в воспалении участвуют молекулярные паттерны, ассоциированные с повреждением (damage-associated molecular pattern (DAMPs)), к которым относят алармины (ядерный белок HMGB1 (high mobility group box 1)), группа кальций-связывающих белков S100, белки теплового шока (heat shock proteins HSP), мочевая и гиалуроновая кислота, фибриноген и др. Индукторы воспаления связываются с белками-рецепторами, такими как TLRs, и активируют биологические реакции в резидентных клетках [23]. Важную роль в инициации воспаления жировой ткани играет нарушение взаимоотношений между адипоцитами и макрофагами [11, 12, 13]. Метаболические нарушения смещают баланс между про- и противовоспалительными регуляторами макрофагов в сторону образования М1-макрофагов и развития дисфункции адипоцитов. Адипоциты страдающих ожирением лиц секретируют провоспалительные цитокины (интерлейкины 1 и 6 (interleukin (IL-1, IL-6)), лептин, фактор некроза опухоли альфа (tumor necrosis factor alpha (TNF-α)), хемокины (моноцитарный хемоаттрактантный белок (monocyte chemoattractant proteins (МСР-1)) и макрофагальный воспалительный белок (macrophage inflammatory protein (MIP-1)), которые способствуют рекрутированию иммунных клеток и активируют воспалительные сигнальные сети [20]. Таким образом, связанное с ожирением хроническое воспаление является важной составляющей патогенеза ИР.
В то же время в ряде работ сообщалось, что нейтрализация провоспалительных путей, управляемых TNFa, IL-1 или IL-6, усугубляет метаболические осложнения [24]. Так, фактор, ассоциированный с рецептором фактора некроза опухолей (Tumor-Necrosis Receptor Associated Factor (TRAF)), может блокировать данные провоспалительные сигналы [25]. Продемонстрировано, что дефицит TRAF-1 предотвращал развитие ожирения путем индукции липолитических путей [26]. Индуцированный воспалением липолиз необходим для высвобождения энергетических ресурсов во время стресса и развития инфекционного процесса. Таким образом, низкоуровневое вялотекущее хроническое воспаление важно для защиты от развития дисфункции адипоцитов [27]. Неалкогольный жировой гепатоз тесно связан с ожирением и МС, однако у некоторых пациентов данное заболевание развивается при отcутствии ожирения [19]. В экспериментальных условиях подтверждено, что высокожировая диета в течение 6 месяцев приводила к развитию ожирения печени и хронического воспаления, характеризующегося повышением уровня TNF-α, IL-13 и IL-18. После 12 месяцев данной диеты наблюдалась гиперэкспрессия IL-6, IL-10, IL-13, циклооксигеназы-2 (COX-2) и TLR. По заключению авторов, данный иммунный ответ оказывает защитный эффект, предотвращая развитие метаболических нарушений [19].
Эти результаты позволяют предположить, что провоспалительная сигнализация не является исключительно патогенной при ожирении. На сегодняшний день остается спорным, действительно ли данный процесс является исключительно неблагоприятным для организма. В связи с этим особенно актуальным является изучение воспалительных сигнальных путей, участвующих в модуляции хронического воспаления жировой ткани, которые практически не раскрыты.
ТОЛЛ-ПОДОБНЫЕ РЕЦЕПТОРЫ
Образ-распознающие рецепторы (pattern recognition receptors (PRRs)) представляют собой белки, локализованные на поверхности клеток иммунной системы, которые способствуют обнаружению молекулярных паттернов, образованных от патогенных микроорганизмов [28]. Функционально данные рецепторы делятся на сигнальные (TLRs) и эндоцитозные (маннозные рецепторы макрофагов). В последнее время TLRs получают все больше внимания, поскольку являются центральными участниками как врожденных, так и адаптивных иммунных реакций [29].
TLRs распознают патогены, локализованные внеклеточно или находящиеся в эндосомах. Идентифицировано 13 TLRs человека (TLRs1–13). Каждый из TLRs отвечает за распознавание определенного набора молекулярных паттернов. После синтеза TLR3, TLR7, TLR8 и TLR9 находятся в эндоплазматическом ретикулуме и переходят в эндолизосомный отдел клетки при ее активации (эндосомальные TLRs), в то время как TLR1, TLR2, TLR4, TLR5, TLR6 и TLR10 локализуются только на плазматической мембране клеток (TLRs, экспрессируемые на клеточной мембране или поверхностные TLRs) [30]. Лигандами для TLRs, экспрессируемых на мембране клетки, служат компоненты микробных мембран, бактериальные протеины и белки вирусов, для эндосомальных TLRs – нуклеиновые кислоты микробов [31]. В распознавание лигандов в наибольшей степени вовлечены TLR2, TLR4, TLR7, TLR8, TLR9. Важной особенностью TLRs является их участие в развитии иммунного ответа на вирусные и бактериальные инфекции [32]. Все эндосомальные TLRs распознают нуклеиновые кислоты микробов, TLR3 распознает двухцепочечную РНК, TLR7 и TLR8 – одноцепочечную РНК, а TLR9 распознает ДНК [33].
Структура TLRs представлена двумя доменами. N-концевая часть TLRs имеет LRR домен (Leucine-Rich Repeat domain), связывающий лиганд. С-концевая часть TLRs имеет TIR домен (Toll/interleukin-1 receptor and Resistance domain), локализована в цитоплазме и взаимодействует с молекулами сигнальных путей [34]. Через TIR сигнал передается на соответствующие киназы, которые активируют факторы транскрипции, ответственные как за индукцию врожденного иммунного ответа посредством экспрессии различных провоспалительных цитокинов и антимикробных факторов, так и за стимуляцию приобретенного иммунного ответа, презентацию антигенов и ряд других процессов [28]. В передаче сигнала задействованы сигнальные пути с участием MyD88 (myeloid differentiation protein 88 (MyD88)), TIRAP (TIR-доменсодержащие адапторы), TICAM1 (TIR-domain-containing adapter-inducing interferon-β (TRIF)) и TICAM2 (TIR-containing adaptеr molecule). Все TLRs (кроме TLR3, передающего сигнал через TRIF) реализуют свое действе посредством сигнального пути MyD88. TLR4 активирует как MyD88-зависимые, так и эндосомальные, TRIF-зависимые сигнальные пути [34].
TLRs широко представлены на поверхности клеток иммунной системы (макрофаги, дендритные, тучные клетки, нейтрофилы, базофилы, В- и Т-клетки, натуральные киллеры) и неиммунных клетках (фибробласты, эпителиальные клетки, кератиноциты) [29]. Кроме того, адипоциты также экспрессируют TLRs, которые активно участвуют не только в антибактериальной и противовирусной защите, но и вовлечены в инициацию хронического воспалительного состояния жировой ткани [35]. Наибольший интерес представляют TLR2, TLR4 и TLR10, экспрессируемые на клеточной мембране, а также TLR9, относящийся к эндосомальным TLRs.
ТОЛЛ-ПОДОБНЫЕ РЕЦЕПТОРЫ В РАЗВИТИИ МЕТАБОЛИЧЕСКОЙ ДИСФУНКЦИИ ПРИ ОЖИРЕНИИ
Несколько исследований продемонстрировали роль TLR2 и TLR4 в распознавании продуктов перекисного окисления липидов, активных форм кислорода, которые участвуют в патогенезе развития ИР и сахарного диабета 2 типа [36]. Окислительный стресс способствует экспрессии провоспалительных цитокинов, включая IFN-γ, IL-1β, IL-6, TNF-α и регулирует экспрессию TLR2/4 через MAPK/NF-kB зависимую сигнализацию (митоген-активированная протеинкиназа МАРК)/ядерный фактор каппа B (nuclear factor-kappa B (NF-kB)), участвуя в развитии метаболического воспаления [35]. Рецептор NLRP3 (nod-like receptor with pyrin domain containing 3) признан центральной воспалительной молекулой в PRR-путях, играющих значимую роль в индукции и прогрессировании воспаления за счет влияния на секрецию различных воспалительных цитокинов [37]. NLRP3 играет ключевую роль в активации TLR, которая зависит от связывания белка MyD88 [38], активирующего NF-kB и митоген-активированный белок. Это свидетельствует о том, что TLR играет важную роль в регулировании воспаления посредством рецептора NLRP3 (рис. 1).
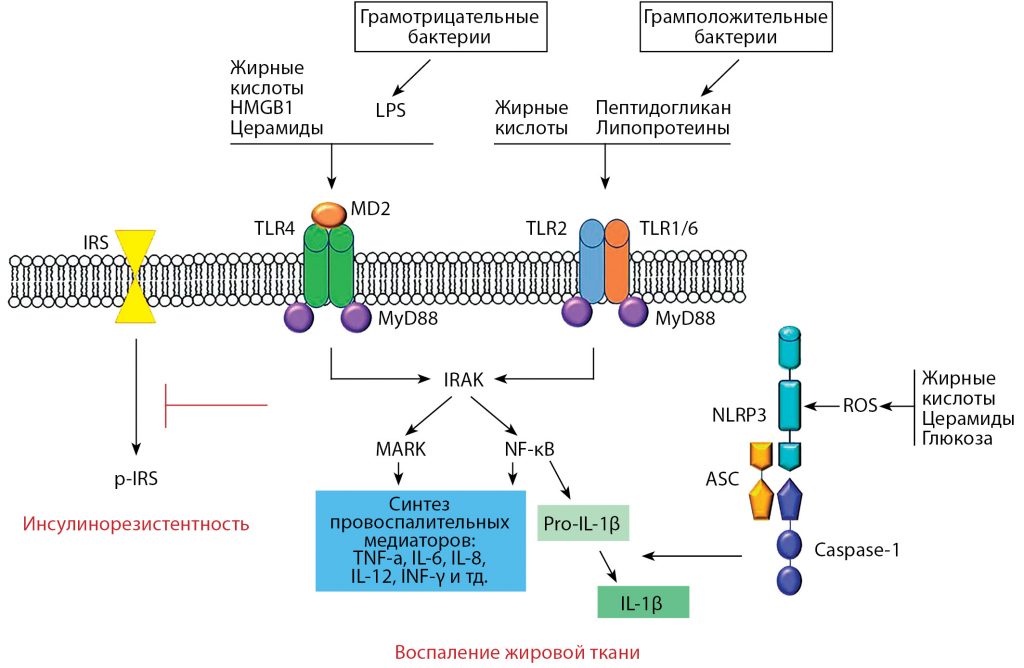
Рисунок 1. Участие толл-подобных рецепторов (TLRs) в сигнальных механизмах воспаления в жировой ткани и инсулинорезистентности
Примечание. Насыщенные жирные кислоты, церамиды, липополисахариды (LPS – от англ. lipopolysaccharide) грамотрицательных бактерий активируют TLR4. Тогда как жирные кислоты, поступающие с пищей, а также пептидогликан и липопротеины грамположительных бактерий активируют TLR2 и TLR1/6. Кроме того, питательные вещества, такие как насыщенные жирные кислоты и их метаболиты, церамиды, могут взаимодействовать с TLR4 и косвенно активировать этот рецептор посредством производства амфотерина или негистонового ядерного белка (HMGB-1, от англ. –high-mobility group box 1). В связывании TLR4 и LPS участвует MD-2 (от англ. – myeloid differentiation factor 2), образуя так называемый TLR4/MD-2 комплекс. После стимуляции лигандами TLRs адапторные молекулы MyD88 (от англ. – myeloid differentiation protein 88) рекрутируют киназу, связанную с рецептором IL-1R (IRAK от англ. – IL-1R-associated kinase), которая, в свою очередь, может активировать либо митоген-активированный протеинкиназой (MAPK от англ. – mitogen-activated protein kinase) сигнальный путь, либо стимулировать ядерный фактор транскрипции kB (NF-kB от англ. – nuclear factor-kappa B). Запуск MAPK или NF-kB сигналинга детерминирует синтез провоспалительных цитокинов (TNF-a, IL-6, IL-8, IL-12, INF-γ и т.д.), вызывающих воспаление жировой ткани. Среди них IL-1β модифицируется в зрелую форму под воздействием комплекса NLRP3 (от англ. – nod-like receptor with pyrin domain containing 3), состоящего из NLRP3, ASC (от англ. – apoptosis-associated speck-like protein) и каспазы-1 (от англ. – caspase-1). Oкислительный стресс (ROS) в ответ на высокий уровень жирных кислот, церамидов или глюкозы может вызвать активацию каспазы-1 комплекса NLRP3, которая, в свою очередь, способствует образованию IL-1β. Воспалительный сигналинг, вызванный LPS или насыщенными жирными кислотами через TLR4/MD-2, может ингибировать фосфорилирование инсулинового рецептора (pIRS, от англ. – phosphorylation of insulin receptor substrate protein (IRS), от англ. – insulin receptor substrate proteins).
Существует большое количество исследований, проведенных в условиях in vitro и связывающих активность TLR4 с развитием ИР [39–44]. Инсулинорезистентные лица имеют повышенный уровень свободных жирных кислот, которые являются лигандами для провоспалительного TLR4 [41]. Данный рецептор экспрессируется моноцитами, играющими ключевую роль в развитии ИР и активирующимися при ожирении и сахарном диабете 2 типа. Инфильтрируя жировую ткань, моноциты становятся тканевыми макрофагами и секретируют хемокины для дальнейшего облегчения рекрутирования иммунных клеток, тем самым потенцируя воспаление. Моноциты и макрофаги являются одними из основных источников цитокинов, таких как TNF-α и IL-1, которые ингибируют инсулиновую сигнализацию посредством активации MAPK и ингибитора kB киназы β (IKKß) для NFkB, который является провоспалительным ядерным транскрипционным фактором. Повышение уровня свободных жирных кислот у лиц с нормальным индексом массы тела стимулирует экспрессию TLR4 и активирует MAPK в циркулирующих моноцитах [42]. Таким образом, провоспалительное состояние, характерное для ожирения и диабета 2 типа, может быть вызвано провоспалительными моноцитами и макрофагами, активированными повышенным уровнем свободных жирных кислот посредством TLRs. Кроме того, экспрессия TLR4 в периферических мононуклеарных клетках снижается в случае потери веса у лиц с избыточной массой тела с МС [43]. Воспаление жировой ткани, опосредованное TLR2/4, играет ключевую роль в активации ренин-ангиотензиновой системы [44]. Известно, что как гиперурикемия, так и ренин-ангиотензиновая система тесно связаны с множественными метаболическими заболеваниями. Мочевая кислота способна регулировать активность ренин-ангиотензиновой системы в адипоцитах. Представленные данные позволяют предположить, что ингибирование TLR4 может подавлять развитие хронического воспаления при ожирении.
Недавние исследования выявили связь между воспалением жировой ткани, связанным с ожирением и повышенной экспрессией TLRs, и семейством Kruppel-подобных факторов транскрипции (Kruppel-like factors (KLFs)), играющим важную роль в дифференцировке адипоцитов [45].
Бактериальные ингредиенты являются одними из главных лигандов TLRs [46]. Так, LPS являются лигандами TLR4, которые присутствуют в клеточной стенке грамотрицательных бактерий. Пептидогликан и липотейхоевая кислота являются лигандами TLR2, присутствуя в клеточной стенке грамположительных бактерий. Очевидно, что микрофлора кишечника человека содержит основные лиганды TLRs. Новые данные демонстрируют, что нарушение кишечной микрофлоры является триггером развития ожирения в результате нарушения сигнализации TLRs [47]. В связи с этим TLRs могут быть терапевтической мишенью у пациентов с ожирением и связанными с ним заболеваниями [48].
Эти исследования подчеркивают способность TLR4 функционировать в качестве метаболического регулятора.
Адипоцитарный TLR9 является предполагаемым новым защитным фактором при воспалении жировой ткани, сопровождающем ожирение [49]. Данный рецептор представлен в разнообразных иммунных клетках. Природными лигандами TLR9 служат участки ДНК бактерий и вирусов [33]. Экспрессия TLR9 значительно повышена в висцеральной жировой ткани у пациентов с недиабетическим ожирением и коррелирует с системными уровнями резистина, что требует дальнейшего изучения.
Недавно противовоспалительные свойства были обнаружены у TLR10, единственного члена семейства TLR с ингибирующей активностью. Показано, что TLR10 влияет на морфологию жировой ткани при ожирении [18]. У людей, страдающих ожирением, у лиц с полиморфизмом гена TLR10 наблюдалось снижение инфильтрации макрофагов в жировой ткани, сопровождающееся тенденцией к снижению уровня лептина и повышению уровня адипонектина в плазме крови. У здоровых лиц с одинаковыми полиморфизмами в гене TLR10 не наблюдалось различий в концентрациях лептина и адипонектина в плазме крови.
Таким образом, TLRs (TLR2, TLR4, TLR9, TLR10) играют значимую роль в патофизиологии ожирения, и их дальнейшее изучение может являться эффективным направлением для подавления хронического воспаления жировой ткани.
Результаты исследований Subramanian S. и соавт. свидетельствуют о потенциальной патофизиологической роли в индуцированной ожирением резистентности к инсулину не только TLRs, но и рецептора конечных продуктов гликирования белков (receptor for advanced glycation end products (RAGE)), триггерного рецептора, экспрессируемого на миелоидных клетках (triggering receptor expressed on myeloid cells (TREM-1)) и амфотерина или негистонового ядерного белка (DNA-binding high-mobility group box 1 (HMGB-1)) [14].
RAGE является трансмембранным гликопротеином типа I, опосредующим различные физиологические функции. Кроме того, данный рецептор является главным медиатором врожденного иммунного ответа, инициирующим развитие воспалительного процесса посредством индукции провоспалительных цитокинов и хемокинов [50]. Продемонстрировано, что RAGE вовлечен в развитие хронического вялотекущего воспаления при метаболических процессах и ИР, индуцированной ожирением [51]. Передача сигналов через RAGE активирует провоспалительный ядерный фактор транскрипции NF-kB, который, в свою очередь, поддерживает экспрессию RAGE. Повреждение тканей при воспалительной реакции сопровождается выработкой лигандов RAGE, которые приводят к RAGE-опосредованному синтезу NF-kB р65 мРНК [50].
Одним из лигандов RAGE является протеин HMGB1 – негистоновый хромосомный белок [52]. Было показано, что связывание HMGB1 с RAGE приводит к снижению концентрации RAGE на поверхности клеток. Являясь важным эндогенным провоспалительным фактором, HMGB1 участвует в патогенезе многих заболеваний. Данный белок является сигнальной молекулой тревоги, вырабатываемой в результате инициации макрофагального ответа на провоспалительные стимулы и при повреждении клеток [53]. Амфотерин относится к молекулярным паттернам, ассоциированным с повреждением (DAMPs). При нарушении функции адипоцитов наблюдается пассивное высвобождение клеточного содержимого, включая HMGB1, во внеклеточную среду. Внеклеточный HMGB1 действует как аллармин, стимулируя активацию резидентных иммунных клеток жировой ткани. Эти клетки секретируют дополнительный HMGB1, который, в свою очередь, активирует/рекрутирует дополнительные иммунные клетки и индуцирует гибель адипоцитов [54]. Во внеклеточном пространстве HMGB1 реализует свое провоспалительное действие через RAGE или TLRs [53]. В результате взаимодействия с ними происходит активация клеток сосудистого эндотелия, макрофагов и моноцитов, экспрессирующих провоспалительные цитокины (IL-β, IL-10, IL-12, TNFa) [55]. HMGB1 способствует высвобождению цитокинов через митоген-активированную протеинкиназу MAPK, ERK1/2 и NF-kB пути. В исследовании Jiang H. и соавт. показано, что существуют тесные взаимоотношения между TLR4, HMGB1 и NF-kB [56]. Так, HMGB1 активирует TLR2 и TLR4, что приводит к инициированию воспаления через MyD88 и NF-kB и высвобождению провоспалительных цитокинов [57]. Таким образом, HMGB1 инициирует молекулярную сигнализацию, которая завершается высвобождением провоспалительных цитокинов и развитием хронического воспаления.
HMGB1 является лигандом не только для RAGE и TLR, но и TREM-1 [14]. Активация TREM-1 – важный механизм хронического воспаления, которое может привести к развитию ИР, индуцированной ожирением. Активация TREM-1 сопровождается повышенной секрецией TNF-α, хемокинов и цитокинов (IL-6, IL-8 и IL-12). При МС наблюдается TREM1-индуцированная активация TLRs в липидных рафтах. Кроме того, TREM-1 взаимодействует не только с TLR, но и с RAGE.
Показано, что пациенты с сахарным диабетом 2 типа, страдающие ожирением, имели более высокую экспрессию TREM-1 в сочетании с HMGB1, RAGE, TLR4 и TLR2 по сравнению с пациентами, страдающими ожирением без диабета [14].
Таким образом, представленные результаты свидетельствуют о потенциальной патофизиологической роли TLRs в индуцированной ожирением резистентности к инсулину, которые тесно взаимосвязаны с другими сигнальными путями.
ЗАКЛЮЧЕНИЕ
Ожирение является актуальной медико-социальной проблемой мирового уровня, требующей решения в связи с распространенностью и глобальными затратами со стороны системы здравоохранения. Жировая ткань – не только место депонирования энергетических субстратов, но и источник секреции провоспалительных и противовоспалительных медиаторов, участвующих в развитии хронического латентного системного воспалительного процесса в организме при ожирении.
Хроническое воспалительное состояние играет ключевую роль в патофизиологии ИР, индуцированной ожирением. Метаболический сигнал при ожирении способствует поляризации макрофагов в М1-направлении и запуске иммунного ответа по Тh1 типу, вызывая хроническое воспаление в жировой ткани. Возможным патофизиологическим звеном развития ИР при воспалении могут быть толл-подобные рецепторы (TLRs), воспринимающие метаболический сигнал. В то же время индуцированный воспалением липолиз необходим для высвобождения энергетических ресурсов во время стресса и развития инфекционного процесса. Таким образом, низкоуровневое вялотекущее хроническое воспаление важно для защиты от дисфункции адипоцитов. Эти результаты позволяют предположить, что провоспалительная сигнализация не является исключительно патогенной при ожирении. На сегодняшний день остается спорным, действительно ли данный процесс является исключительно неблагоприятным для организма. В связи с этим особенно актуальным является изучение воспалительных сигнальных путей, участвующих в модуляции хронического воспаления жировой ткани.
Очевидно, что дальнейшее изучение воспалительных сигнальных путей с участием TLRs, инициирующих развитие хронического воспаления жировой ткани, позволит разработать новые и эффективные терапевтические стратегии в отношении ожирения и его метаболических осложнений.
ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Источник финансирования. Подготовка и публикация рукописи проведены на личные средства авторского коллектива.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Участие авторов. Все авторы внесли значимый вклад в проведение исследования и подготовку статьи, прочли и одобрили финальную версию статьи перед публикацией.
Список литературы
↑1. Moore JB, Boesch C. Getting energy balance right in an obesogenic world. Proc Nutr Soc. 2019;78(3):259-261. https://doi.org/10.1017/S0029665118002720
↑2. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet. 2016;387(10026):1377-1396. https://doi.org/10.1016/s0140-6736(16)30054-x
↑3. Ожирение в России: статистические данные. 2013. [Ozhirenie v Rossii: statisticheskie dannye. 2013. (In Russ.)]
↑4. Bhupathiraju SN, Hu FB. Epidemiology of Obesity and Diabetes and Their Cardiovascular Complications. Circ Res. 2016;118(11):1723-1735. https://doi.org/10.1161/CIRCRESAHA.115.306825
↑5. Кытикова О.Ю., Антонюк М.В., Гвозденко Т.А., Новгородцева Т.П. Метаболические аспекты взаимосвязи ожирения и бронхиальной астмы. // Ожирение и метаболизм. – 2018. – Т. 15. – №4. – С. 9-14. [Kytikova OY, Antonyuk MV, Gvozdenko TA, Novgorodtseva TP. Metabolic aspects of the relationship of asthma and obesity. Obesity and metabolism. 2018;15(4):9-14. (In Russ.)] https://doi.org/10.14341/OMET9578
↑6. Collaborators GBDEMRO. Burden of obesity in the Eastern Mediterranean Region: findings from the Global Burden of Disease 2015 study. Int J Public Health. 2018;63(Suppl 1):165-176. https://doi.org/10.1007/s00038-017-1002-5
↑7. DeBoer MD. Assessing and Managing the Metabolic Syndrome in Children and Adolescents. Nutrients. 2019;11(8). https://doi.org/10.3390/nu11081788
↑8. Kumari R, Kumar S, Kant R. An update on metabolic syndrome: Metabolic risk markers and adipokines in the development of metabolic syndrome. Diabetes Metab Syndr. 2019;13(4):2409-2417. https://doi.org/10.1016/j.dsx.2019.06.005
↑9. Gruss SM, Nhim K, Gregg E, et al. Public Health Approaches to Type 2 Diabetes Prevention: the US National Diabetes Prevention Program and Beyond. Curr Diab Rep. 2019;19(9):78. https://doi.org/10.1007/s11892-019-1200-z
↑10. Zhukova NV, Novgorodtseva TP, Denisenko YK. Effect of the prolonged high-fat diet on the fatty acid metabolism in rat blood and liver. Lipids Health Dis. 2014;13:49. https://doi.org/10.1186/1476-511X-13-49
↑11. Huh JY, Park YJ, Ham M, Kim JB. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol Cells. 2014;37(5):365-371. https://doi.org/10.14348/molcells.2014.0074
↑12. Revelo XS, Tsai S, Lei H, et al. Perforin is a novel immune regulator of obesity-related insulin resistance. Diabetes. 2015;64(1):90-103. https://doi.org/10.2337/db13-1524
↑13. Thomas D, Apovian C. Macrophage functions in lean and obese adipose tissue. Metabolism. 2017;72:120-143. https://doi.org/10.1016/j.metabol.2017.04.005
↑14. Subramanian S, Pallati PK, Sharma P, et al. Significant association of TREM-1 with HMGB1, TLRs and RAGE in the pathogenesis of insulin resistance in obese diabetic populations. Am J Transl Res. 2017;9(7):3224-3244.
↑15. Wong SK, Chin KY, Ima-Nirwana S. Toll-like Receptor as a Molecular Link between Metabolic Syndrome and Inflammation: A Review. Curr Drug Targets. 2019;20(12):1264-1280. https://doi.org/10.2174/1389450120666190405172524.
↑16. Liu YC, Zou XB, Chai YF, Yao YM. Macrophage polarization in inflammatory diseases. Int J Biol Sci. 2014;10(5):520-529. https://doi.org/10.7150/ijbs.8879.
↑17. Follow-up to the Political Declaration of the High-level Meeting of the General Assembly on the Prevention and Control of Non-Communicable Diseases. Geneva: World Health Assembly; 2013.
↑18. Boutens L, Mirea AM, van den Munckhof I, et al. A role for TLR10 in obesity and adipose tissue morphology. Cytokine. 2018;108:205-212. https://doi.org/10.1016/j.cyto.2018.03.021
↑19. Sigrist-Flores SC, Ponciano-Gomez A, Pedroza-Gonzalez A, et al. Chronic intake of moderate fat-enriched diet induces fatty liver and low-grade inflammation without obesity in rabbits. Chem Biol Interact. 2019;300:56-62. https://doi.org/10.1016/j.cbi.2019.01.004
↑20. Kane H, Lynch L. Innate Immune Control of Adipose Tissue Homeostasis. Trends Immunol. 2019;40(9):857-872. https://doi.org/10.1016/j.it.2019.07.006
↑21. Lin TY, Chiu CJ, Kuan CH. et Lin TY, Chiu CJ, Kuan CH, et al. IL-29 promoted obesity-induced inflammation and insulin resistance. Cell Mol Immunol. 2020;17(4):369-379. https://doi.org/10.1038/s41423-019-0262-9
↑22. Krejsek J. Defensive and damaging inflammation: basic characteristics. Vnitr Lek. 2019;65(2):76-80.
↑23. Zielen S, Trischler J, Schubert R. Lipopolysaccharide challenge: immunological effects and safety in humans. Expert Rev Clin Immunol. 2015;11(3):409-418. https://doi.org/10.1586/1744666X.2015.1012158
↑24. Stanley TL, Zanni MV, Johnsen S, et al. TNF-alpha antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J Clin Endocrinol Metab. 2011;96(1):E146-150. https://doi.org/10.1210/jc.2010-1170
↑25. Anto Michel N, Colberg C, Buscher K, et al. Inflammatory Pathways Regulated by Tumor Necrosis Receptor-Associated Factor 1 Protect From Metabolic Consequences in Diet-Induced Obesity. Circ Res. 2018;122(5):693-700. https://doi.org/10.1161/CIRCRESAHA.117.312055
↑26. Missiou A, Kostlin N, Varo N, et al. TRAF1 Deficiency Attenuates Atherosclerosis in Mice by Impairing Monocyte Recruitment to the Vessel Wall. Circulation. 2010;121(18):2033-2044. https://doi.org/10.1161/CIRCULATIONAHA.109.895037
↑27. Wernstedt Asterholm I, Tao C, Morley TS, et al. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014;20(1):103-118. https://doi.org/10.1016/j.cmet.2014.05.005
↑28. Wang X, Zhang Y, Zhang R, Zhang J. The diversity of pattern recognition receptors (PRRs) involved with insect defense against pathogens. Curr Opin Insect Sci. 2019;33:105-110. https://doi.org/10.1016/j.cois.2019.05.004
↑29. Zakeri A, Russo M. Dual Role of Toll-like Receptors in Human and Experimental Asthma Models. Front Immunol. 2018;9:1027. https://doi.org/10.3389/fimmu.2018.01027
↑30. De Nardo D. Toll-like receptors: Activation, signalling and transcriptional modulation. Cytokine. 2015;74(2):181-189. https://doi.org/10.1016/j.cyto.2015.02.025
↑31. Schaefer L. Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem. 2014;289(51):35237-35245. https://doi.org/10.1074/jbc.R114.619304
↑32. Ashour DS. Toll-like receptor signaling in parasitic infections. Expert Rev Clin Immunol. 2015;11(6):771-780. https://doi.org/10.1586/1744666X.2015.1037286
↑33. Lee BL, Barton GM. Trafficking of endosomal Toll-like receptors. Trends Cell Biol. 2014;24(6):360-369. https://doi.org/10.1016/j.tcb.2013.12.002
↑34. Mirotti L, Alberca Custodio RW, Gomes E, et al. CpG-ODN Shapes Alum Adjuvant Activity Signaling via MyD88 and IL-10. Front Immunol. 2017;8:47. https://doi.org/10.3389/fimmu.2017.00047
↑35. Akhter N, Madhoun A, Arefanian H, et al. Oxidative Stress Induces Expression of the Toll-Like Receptors (TLRs) 2 and 4 in the Human Peripheral Blood Mononuclear Cells: Implications for Metabolic Inflammation. Cell Physiol Biochem. 2019;53(1):1-18. https://doi.org/10.33594/000000117
↑36. Andrews M, Soto N, Arredondo-Olguin M. Association between ferritin and hepcidin levels and inflammatory status in patients with type 2 diabetes mellitus and obesity. Nutrition. 2015;31(1):51-57. https://doi.org/10.1016/j.nut.2014.04.019
↑37. Gong T, Yang Y, Jin T, et al. Orchestration of NLRP3 Inflammasome Activation by Ion Fluxes. Trends Immunol. 2018;39(5):393-406. https://doi.org/10.1016/j.it.2018.01.009
↑38. Koppenol-Raab M, Sjoelund V, Manes NP, et al. Proteome and Secretome Analysis Reveals Differential Post-transcriptional Regulation of Toll-like Receptor Responses. Mol Cell Proteomics. 2017;16(4 suppl 1):S172-S186. https://doi.org/10.1074/mcp.M116.064261
↑39. Hussey SE, Liang H, Costford SR, et al. TAK-242, a small-molecule inhibitor of Toll-like receptor 4 signalling, unveils similarities and differences in lipopolysaccharide- and lipid-induced inflammation and insulin resistance in muscle cells. Biosci Rep. 2013;33(1):37-47. https://doi.org/10.1042/BSR20120098
↑40. Ieronymaki E, Daskalaki MG, Lyroni K, Tsatsanis C. Insulin Signaling and Insulin Resistance Facilitate Trained Immunity in Macrophages Through Metabolic and Epigenetic Changes. Front Immunol. 2019;10:1330. https://doi.org/10.3389/fimmu.2019.01330
↑41. Reyna SM, Tantiwong P, Cersosimo E, et al. Short-term exercise training improves insulin sensitivity but does not inhibit inflammatory pathways in immune cells from insulin-resistant subjects. J Diabetes Res. 2013;2013:107805. https://doi.org/10.1155/2013/107805
↑42. Liang H, Lum H, Alvarez A, et al. A low dose lipid infusion is sufficient to induce insulin resistance and a pro-inflammatory response in human subjects. PLoS One. 2018;13(4):e0195810. https://doi.org/10.1371/journal.pone.0195810
↑43. de Mello VD, Kolehmainen M, Pulkkinen L, et al. Downregulation of genes involved in NFkappaB activation in peripheral blood mononuclear cells after weight loss is associated with the improvement of insulin sensitivity in individuals with the metabolic syndrome: the GENOBIN study. Diabetologia. 2008;51(11):2060-2067. https://doi.org/10.1007/s00125-008-1132-7
↑44. Zhang J, Diao B, Lin X, et al. TLR2 and TLR4 mediate an activation of adipose tissue renin-angiotensin system induced by uric acid. Biochimie. 2019;162:125-133. https://doi.org/10.1016/j.biochi.2019.04.013
↑45. Wang C, Ha X, Li W, et al. Correlation of TLR4 and KLF7 in Inflammation Induced by Obesity. Inflammation. 2017;40(1):42-51. https://doi.org/10.1007/s10753-016-0450-z
↑46. Eisenbarth SC, Piggott DA, Huleatt JW, et al. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196(12):1645-1651. https://doi.org/10.1084/jem.20021340
↑47. Bahadur T, Chaudhry R, Bamola VD, et al. Toll like receptors (TLRs) in response to human gut microbiota of Indian obese and lean individuals. J Family Med Prim Care. 2019;8(5):1567-1570. https://doi.org/10.4103/jfmpc.jfmpc_136_19
↑48. Miura K, Ishioka M, Iijima K. The Roles of the Gut Microbiota and Toll-like Receptors in Obesity and Nonalcoholic Fatty Liver Disease. J Obes Metab Syndr. 2017;26(2):86-96. https://doi.org/10.7570/jomes.2017.26.2.86
↑49. Thomalla M, Schmid A, Neumann E, et al. Evidence of an anti-inflammatory toll-like receptor 9 (TLR 9) pathway in adipocytes. J Endocrinol. 2019;240(2):325-343. https://doi.org/10.1530/JOE-18-0326
↑50. Kierdorf K, Fritz G. RAGE regulation and signaling in inflammation and beyond. J Leukoc Biol. 2013;94(1):55-68. https://doi.org/10.1189/jlb.1012519
↑51. Song F, Hurtado del Pozo C, Rosario R, et al. RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes. 2014;63(6):1948-1965. https://doi.org/10.2337/db13-1636
↑52. Paudel YN, Angelopoulou E, Piperi C, et al. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur J Pharmacol. 2019;858:172487. https://doi.org/10.1016/j.ejphar.2019.172487
↑53. Brencicova E, Diebold SS. Nucleic acids and endosomal pattern recognition: how to tell friend from foe? Front Cell Infect Microbiol. 2013;3:37. https://doi.org/10.3389/fcimb.2013.00037
↑54. Zhang J, Zhang L, Zhang S, et al. HMGB1, an innate alarmin, plays a critical role in chronic inflammation of adipose tissue in obesity. Mol Cell Endocrinol. 2017;454:103-111. https://doi.org/10.1016/j.mce.2017.06.012
↑55. Tadié J-M, Bae H-B, Banerjee S, et al. Differential activation of RAGE by HMGB1 modulates neutrophil-associated NADPH oxidase activity and bacterial killing. Am J Physiol Cell Physiol. 2012;302(1):C249-C256. https://doi.org/10.1152/ajpcell.00302.2011
↑56. Jiang H, Duan J, Xu K, Zhang W. Resveratrol protects against asthma-induced airway inflammation and remodeling by inhibiting the HMGB1/TLR4/NF-κB pathway. Exp Ther Med. 2019. https://doi.org/10.3892/etm.2019.7594
↑57. Szczepanski MJ, Luczak M, Olszewska E, et al. Molecular signaling of the HMGB1/RAGE axis contributes to cholesteatoma pathogenesis. J Mol Med. 2014;93(3):305-314. https://doi.org/10.1007/s00109-014-1217-3