Актуальность
Эндогенный гиперкортицизм проявляется поражением многих органов и тканей, обусловленным длительным воздействием на организм избыточного количества кортикостероидов [1]. В этиологии эндогенного гиперкортицизма преобладает АКТГ-зависимый гиперкортицизм (болезнь Иценко–Кушинга – 60–70% случаев, АКТГ-эктопический синдром – 5–10%), тогда как на долю первичного поражения надпочечников (синдром Иценко–Кушинга, СИК) приходится 20–30% случаев [2]. В структуре СИК выделяют одностороннее [аденома надпочечника (75–90%) или адренокортикальный рак (<5%)] и двустороннее поражение надпочечников (10%) [3]. Двустороннюю гиперплазию надпочечников (ДГН) подразделяют в зависимости от размера узлов (макронодулярная при размере узлов более 1 см и микронодулярная), а также по наличию или отсутствию пигментации при морфологическом исследовании. Среди вариантов ДГН выделяют: первичную двустороннюю макронодулярную гиперплазию надпочечников (ПДМГН, primary bilateral macronodular adrenal hyperplasia, PBMAH), первичное пигментное нодулярное поражение надпочечников (primary pigmented nodular adrenocortical disease, PPNAD) и изолированное микронодулярное поражение надпочечников (isolated micronodular adrenocortical disease, iMAD) [4].
Сигнальный путь цАМФ – протеинкиназа А (ПКА) играет важную роль в развитии, пролиферации и функции клеток коры надпочечников. Нарушения в различных компонентах этого сигнального пути могут приводить к развитию СИК. Среди редких синдромов, связанных с мутациями в компонентах цАМФ–ПКА сигнального пути, выделяют: синдром Мак-Кьюн–Олбрайта (постзиготические активирующие мутации в гене GNAS, кодирующем Gsα субъединицу), в рамках которого развивается первичное биморфное заболевание коры надпочечников (primary bimorphic adrenocortical disease); Карни комплекс (герминальные инактивирующие мутации в гене PRKAR1A, кодирующем регуляторную R1α субъединицу ПКА) с возможным развитием PPNAD. Кроме того, инактивирующие герминальные мутации в генах фосфодиэстераз (ферментов, связывающих и инактивирующих цАМФ) (PDE11A и PDE8B) были описаны как при ПДМГН, так и при PPNAD и iMAD [4]. Соматические активирующие мутации в гене, кодирующем каталитическую субъединицу ПКА (PRKACA), были обнаружены в 40% кортикостером [5]. В последнее десятилетие описано несколько пациентов с ДГН и СИК с герминальным увеличением числа копий гена PRKACA [5]. Также был описан единичный случай активирующей мутации в гене рецептора АКТГ (MC2R) у пациента с ПДМГН [6].
При ПДМГН выявлены генетические дефекты, ассоциированные с другими сигнальными путями, помимо цАМФ–ПКА. ПДМГН крайне редко может быть компонентом синдрома множественных эндокринных неоплазий 1 типа, обусловленного мутациями в гене MEN1; семейного аденоматозного полипоза, обусловленного мутациями в гене APC; наследственного лейомиоматоза и почечно-клеточного рака [hereditary leiomyomatosis and renal cell carcinoma (HLRCC), обусловленного мутациями в гене фумаратгидратазы (FH) [5].
В 2013 г. G. Assie и соавт. выявили новый ген, ответственный за развитие СИК вследствие ПДМГН. При исследовании 33 пациентов с ПДМГН и СИК, мутации в гене ARMC5 были обнаружены в 18 случаях (55%) [7]. Во всех узлах выявлялись герминальные мутации в гене, при этом «выключение» второго аллеля различалось в разных узлах: соматические мутации были выявлены в 68% случаев, потеря гетерозиготности (LOH) – в 32% [7], что свидетельствует о том, что ARMC5 является геном-супрессором опухолевого роста. В двух других исследованиях было показано, что мутации в гене ARMC5 встречаются в 25% случаев ПДМГН [8, 9]. Этот ген расположен на хромосоме 16p11.2 и кодирует белок Armadillo repeat containing 5, функции которого не изучены [4]. Влияние на апоптоз миссенс-мутаций в гене ARMC5 изучалось на линиях клеток адренокортикального рака H295R [10]. Было показано, что инактивирующие мутации в гене ARMC5 приводят к утрате способности клеток коры надпочечников индуцировать апоптоз, что может объяснять крупные размеры надпочечников при ПДМГН [10].
Примечательно, что гиперсекреция кортизола при ПДМГН не является истинно АКТГ-независимой, поскольку опухолевые клетки продуцируют АКТГ, который оказывает пара- и аутокринное воздействие на секрецию кортизола [11].
Таким образом, ПДМГН – редкая, генетически гетерогенная причина развития эндогенного гиперкортицизма, в 25–55% случаев обусловленная инактивирующими мутациями в гене ARMC5. Ниже описан случай наследственного СИК в результате ПДМГН, обусловленного мутацией в гене ARMC5.
Описание случая
Пациентка Ю., 37 лет, впервые поступила в отделение нейроэндокринологии и остеопатий ФГБУ «НМИЦ эндокринологии» Минздрава России в сентябре 2016 г. с жалобами на избыточный вес, повышение АД до 160/110 мм. рт.ст., нечистоту кожи, нерегулярный менструальный цикл. Вышеперечисленные жалобы возникли около трех лет назад. При обследовании по месту жительства летом 2016 г. заподозрен эндогенный гиперкортицизм. Результаты лабораторного обследования представлены в таблице. По данным УЗИ, в проекции правого надпочечника определяется округлое гипоэхогенное неоднородное образование 64×32 мм с неровными, полициклическими контурами. При КТ выявлены объемные образования обоих надпочечников кистозно-аденоматозного характера. При МРТ головного мозга без контраста в структуре аденогипофиза выявлен гипоинтенсивный участок диаметром до 0,6 см (МРТ-картина микроаденомы гипофиза).
Результаты лабораторной диагностики на этапах лечения
| Название теста | По месту жительства летом 2016 г. | В ФГБУ «НМИЦ эндокринологии» Минздрава России в сентябре 2016 г. | Референсный интервал |
| АКТГ в 8:00, пг/мл | 0,99 | 1 | 7–66 |
| АКТГ в 23:00, пг/мл | – | 1 | 0–30 |
| Кортизол в 08:00, нмоль/л | – | 1399 | 123–626 |
| Кортизол в 23:00, нмоль/л | – | 1427 | 46–270 |
| Свободный кортизол в суточной моче, нмоль/сут | 1579,6 | 5063,5 | 60–413 |
| Свободный кортизол в слюне в 23:00, нмоль/л | – | 56,6 | 0,5–9,4 |
| Кортизол на фоне ночного подавляющего теста с 1 мг дексаметазона, нмоль/л | 513 | – | <50 |
Гормональный анализ по месту жительства: альдостерон – 80 пг/мл (норма до 392), ренин 10,4 мкМЕд/мл (до 46,1), метанефрин в плазме 16,3 пг/мл (до 30), норметанефрин в плазме 98,2 пг/мл (до 190). Также отмечалось повышение уровня ПТГ до 7,79 пмоль/л (до 6,9).
Мать пациентки в 2003 г. в 45-летнем возрасте проходила обследование в отделении нейроэндокринологии ЭНЦ РАМН, где у нее был диагностирован СИК тяжелого течения. По данным выписного эпикриза, на фоне приема аминоглутетимида (500 мг/сут) уровень АКТГ в 08:00 – 8,4 пг/мл, в 23:00 – 9,5 пг/л; уровень кортизола на фоне ночного подавляющего теста с 1 мг дексаметазона – 249 нмоль/л. При УЗИ – множественные опухоли обоих надпочечников, МРТ-картина с наибольшей вероятностью соответствовала макронодулярной гиперплазии надпочечников. В декабре 2003 г. была выполнена левосторонняя адреналэктомия, в феврале 2004 г. – правосторонняя адреналэктомия.
При обследовании в отделении нейроэндокринологии и остеопатий в сентябре 2016 г. у пациентки Ю. был подтвержден АКТГ-независимый эндогенный гиперкортицизм (см. таблицу). В биохимическом анализе крови: калий – 4,5 ммоль/л (3,5–5,1), глюкоза – 4,42 ммоль/л, холестерин – 7,8 ммоль/л (3,3–5,2). В суточной моче метанефрин – 194 мкг (25–312), норметанефрин – 347 мкг (35–445). По данным МСКТ: надпочечники обычно расположены, с четкими неровными контурами. Оба надпочечника деформированы, неравномерно утолщены. Структура неоднородная за счет многочисленных объемных образований округлой и овальной формы, размером от 12 до 36 мм. Наиболее крупные образования расположены в латеральной ножке левого надпочечника (до 33 мм) и в латеральной ножке правого надпочечника (до 28 и 36 мм). Контуры образований четкие, ровные. Структура однородная. Плотность их по фазам составляет: нативная фаза – артериальная фаза – венозная фаза – отсроченная фаза: 13–37–74–38 ед.Н. Размер правого надпочечника (медиальная ножка–латеральная ножка–тело): 12–20–14 мм. Размер левого надпочечника: 18–22–10 мм. Длина правого надпочечника, включая ножки и тело, – 9,3 см, левого – 8,9 см. (рис. 1, 2). При повторной МРТ головного мозга данных за микроаденому гипофиза не получено. Таким образом, был верифицирован диагноз «Синдром Иценко–Кушинга. Первичная двусторонняя макронодулярная гиперплазия надпочечников». Пациентка была консультирована хирургом, показано хирургическое лечение. Из осложнений эндогенного гиперкортицизма подтверждена артериальная гипертензия 2 степени, по поводу чего назначены лозартан, индапамид, доксазозин с достижением целевых показателей АД.
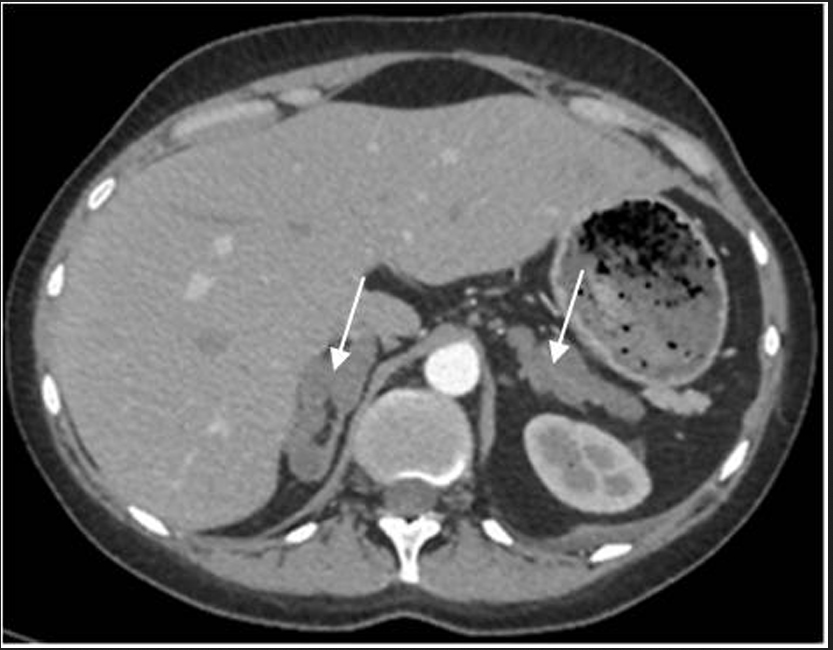
Рис. 1. МСКТ надпочечников пациентки Ю., артериальная фаза, аксиальная проекция (надпочечники указаны стрелками).
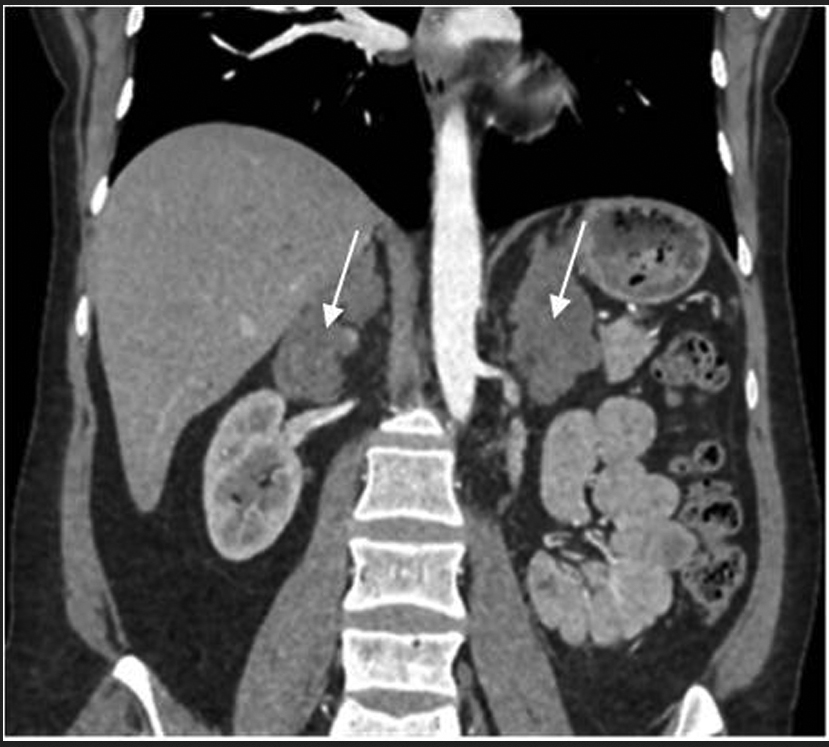
Рис. 2. МСКТ надпочечников пациентки Ю., артериальная фаза, коронарная проекция (надпочечники указаны стрелками).
В связи с анамнестическими данными о повышении уровня ПТГ был исследован уровень общего кальция и выявлено его повышение [2,56 ммоль/л (2,15–2,55)]. При повторном исследовании: кальций общий – 2,61 ммоль/л, кальций ионизированный – 1,24 ммоль/л (1,03–1,29). Уровень ПТГ также оказался повышенным [70,44 пг/мл (15–65)], что подтверждало наличие ПТГ-зависимой гиперкальциемии. При УЗИ выявлены признаки гиперплазии правой верхней околощитовидной железы 0,8×0,5×0,3 см. Учитывая отсутствие нефролитиаза (по данным УЗИ) и снижения минеральной плотности костей (по данным денситометрии), сформулирован диагноз «Первичный гиперпаратиреоз, мягкая форма. Аденома правой верхней околощитовидной железы». В выписном эпикризе матери пациентки от 2003 г. имелись данные об уровне ионизированного кальция – 1,24 ммоль/л (1,03–1,29), однако сведения об уровне общего кальция и ПТГ отсутствовали.
В отделении хирургии ФГБУ «НМИЦ эндокринологии» пациентке Ю. проведена люмболапаротомия слева, левосторонняя адреналэктомия с опухолью, спустя неделю – торакофренолапаротомия справа, правосторонняя адреналэктомия с опухолью. В послеоперационном периоде назначена заместительная терапия гидрокортизоном (суммарная доза 40 мг/сут) и флудрокортизоном (0,05 мг/сут). Послеоперационный период протекал без осложнений. При макроскопическом исследовании: правый надпочечник весом 57,0 г, размером 10,5×5,5×3,0 см, левый – весом 77,0 г, размером 7,0×10,0×3,5 см. Поверхность их крупнобугристая. На разрезе оба представлены охряно-желтыми узлами диаметром от 0,5 до 3,0 см (рис. 3, см. раздел дополнительная информация). Микроскопическая картина соответствует макронодулярной гиперплазии надпочечников. Узлы частью инкапсулированы с соединительнотканными трабекулами, имеют однотипное строение (из клеток, аналогичных кортикальным эндокриноцитам пучковой зоны, сливающихся в крупные тяжи и скопления) (рис. 4, см. раздел дополнительная информация).
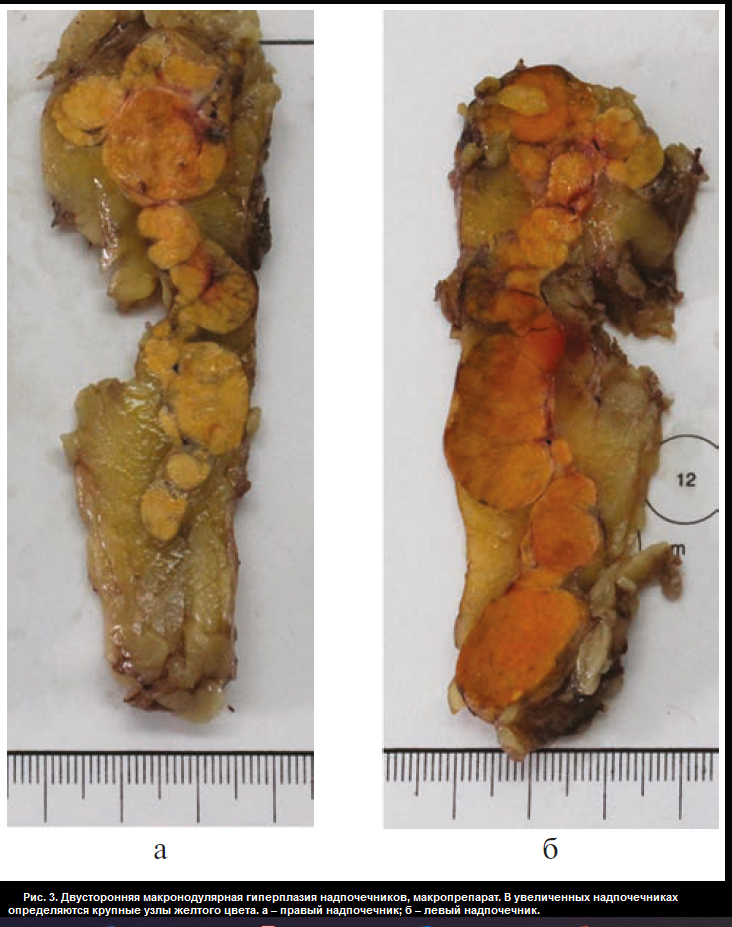
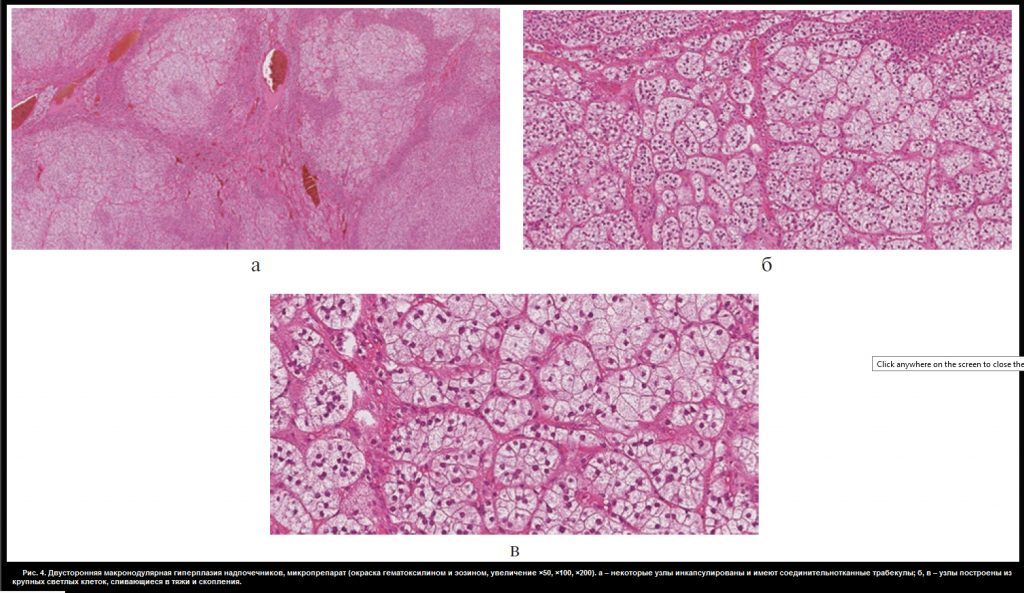
Рис. 4. Двусторонняя макронодулярная гиперплазия надпочечников, микропрепарат (окраска гематоксилином и эозином, увеличение ×50, ×100, ×200). а – некоторые узлы инкапсулированы и имеют соединительнотканные трабекулы; б, в – узлы построены из крупных светлых клеток, сливающиеся в тяжи и скопления.
При повторной госпитализации в отделение нейроэндокринологии и остеопатий в феврале 2018 г. диагностирована медикаментозная компенсация хронической надпочечниковой недостаточности на фоне приема гидрокортизона в суточной дозе 30 мг, флудрокортизона в дозе 0,05 мг. После операции у пациентки нормализовались менструальный цикл и АД, снизился вес на 12 кг. Как при амбулаторном обследовании, так и во время повторной госпитализации, сохранялась гиперкальциемия (кальций общий – 2,68–2,78 ммоль/л, кальций ионизированный – 1,22–1,28 ммоль/л) при уровне ПТГ ближе к верхней границе нормы – 54,95 пг/мл. По данным УЗИ, сохранялась гиперплазия правой верхней околощитовидной железы. Рекомендовано динамическое наблюдение.
Учитывая семейный анамнез (наличие СИК как следствие ПДМГН у матери), для уточнения генетической причины заболевания было проведено секвенирование экзома. Геномную ДНК из лимфоцитов периферической крови выделяли с помощью набора PureLink Genomic DNA Mini Kit, согласно инструкциям производителя (Thermo Fisher Scientific, США). Ультразвуковая фрагментация ДНК проводилась на приборе Covaris S220 (Covaris, США). Пробоподготовка библиотек для секвенирования и экзомное обогащение выполнены с использованием набора TruSeq DNA Exome, согласно инструкциям производителя (Illumina, США). Парно-концевое чтение полученных библиотек (2×80 п.н.) проводилось на секвенаторе NextSeq550 (Illumina, США). Для первичного биоинформатического анализа данных использованы программные инструменты bwa, Picard, Genome Analysis ToolKit (GATK). Аннотация выявленных вариантов проводилась с помощью программного пакета ANNOVAR [12]. Выявлен герминальный гетерозиготный вариант в экзоне 6 гена ARMC5 p.R898W (c.2692C>T, референсная последовательность NM_001105247), предсказанный патогенным по in silico алгоритмами SIFT, POLYPHEN2-HDIV, POLYPHE2-HVAR, Mutationtaster. Частота аллеля в популяции, по данным Genome Aggregation Database (http://gnomad.broadinstitute.org), 0,000009825.
Обсуждение
ПДМГН, приводящую к развитию СИК, обычно диагностируют на пятом или шестом десятилетии жизни. Среди пациентов с ПДМГН преобладают женщины, что характерно и для других форм СИК. Истинная распространенность ПДМГН в популяции неизвестна [5]. В представленном нами случае ПДМГН была диагностирована в возрасте 37 лет у пробанда и в возрасте 45 лет у матери, что согласуется с данными литературы. S. Espiard и соавт. сравнивали пациентов с СИК и ПДМГН с мутациями и без мутаций в гене ARMC5. У пациентов с мутациями в этом гене при морфологическом исследовании выявлялось значимо большее количество узлов в надпочечниках, вес надпочечников был существенно больше [10]. У пациентов с мутациями в ARMC5 чаще выявлялся манифестный, а не субклинический СИК, был ниже уровень АКТГ, выше вечерний уровень кортизола в крови, свободного кортизола в суточной моче и подъем содержания кортизола на фоне ночного подавляющего теста с дексаметазоном [10]. Учитывая данные о влиянии мутаций в гене ARMC5 на апоптоз, предполагается, что гиперплазия надпочечников и формирование узлов происходят прогрессивно, но медленно ввиду доброкачественности опухолевого процесса при ПДМГН. Когда масса надпочечников достигает достаточной критической величины, АКТГ-независимый синтез кортизола приводит к явному эндогенному гиперкортицизму. Это может объяснять тот факт, что СИК при ПДМГН развивается достаточно поздно (в зрелом возрасте) [5].
ПДМГН является генетически гетерогенным заболеванием. Гены, мутации в которых выявлены при ПДМГН, включают: ARMC5, MEN1, FH, PDE11A, PDE8B, GNAS, APC, MC2R, PRKACA [4]. До открытия гена ARMC5 мутации в остальных генах объясняли развитие лишь единичных случаев. Исследования последних лет показали, что наиболее частой причиной развития ПДМГН являются инактивирующие мутации в гене ARMC5, кодирующем белок Armadillo repeat-containing 5. К 2015 г. в литературе было описано 29 герминальных и 32 соматических мутации в гене ARMC5 [5]. Среди герминальных мутаций преобладали миссенс-мутации (44,8%); нонсенс-мутации встречались в 24,1%, мутации со сдвигом рамки считывания – в 27,6%, делеции – в 3,4% случаев [5]. Некоторые из этих мутаций были найдены в нескольких неродственных случаях: p.I58Nfs44* [8, 9], p.R267X [7, 8, 10], p.R593W [8, 13], p.R898W [7, 8, 10] и p.A106Rfs31* [10, 14]. В целом мутации в гене ARMC5 распределены равномерно по всей нуклеотидной последовательности, и в настоящее время «горячих точек» для мутаций не выявлено. Найденная нами мутация была описана ранее в литературе [7, 8, 10]. В России мутация в гене ARMC5 описывается впервые.
У нашей пациентки был выявлен также первичный гиперпаратиреоз, что клинически могло соответствовать синдрому МЭН 1 типа. Однако при секвенировании экзома патогенных вариантов в гене MEN1 выявлено не было. В литературе описаны случаи менингиом у пациентов с мутациями в гене ARMC5 [9, 14], однако данные о развитии аденом околощитовидных желез у таких пациентов нами найдены не были. Вопрос о роли мутаций в гене ARMC5 в развитии опухолей околощитовидных желез остается открытым.
Заключение
ПДМГН является редкой причиной развития эндогенного гиперкортицизма. Наиболее часто при ПДМГН выявляют инактивирующие мутации в гене-супрессоре опухолевого роста ARMC5, тем не менее, около 1/2–3/4 пациентов с СИК и ПДМГН, не отобранные по тяжести СИК или по наличию или отсутствию отягощенного семейного анамнеза, не имеют мутаций в известных генах. Применение полноэкзомного секвенирования может быть оправдано при ПДМГН ввиду генетической гетерогенности заболевания. Могут ли мутации в гене ARMC5 предрасполагать к развитию других опухолей, еще предстоит выяснить.